 2019, Vol. 30
2019, Vol. 30 


b University of Chinese Academy of Sciences, Beijing 100049, China
Carfilzomib is a proteasome inhibitor for the treatment of multiple myeloma (MM) which is the second common type of malignancy and a progressive B-cell tumour [1-3]. In 2012, carfilzomib (marketed under the brand name Kyprolis) was approved as cancer drug by the U.S. Food and Drug Administration (FDA) and then followed by Europe in 2015, and Japan in 2016. As shown in Fig. 1, carfilzomib derived from epoxomicin and YU-101 is made up of a tetrapeptide, and the elimilation of three chiral centers from epoxomicin to carfilzomib greatly simplifies the synthetic routes [4]. Apart from the original medicinal chemistry route developed by Crews group and Proteolix, much more efforts have been taken to exploit potential manufacturing and alternate routes to carfilzomib [5-10]. Among these synthetic routes, the last step always involved the reaction of a tripeptide with the epoxyketone fragment (as marked in Fig. 1). Presently, the synthesis of epoxyketone remained to be the major challenge, and when using the vinyl ketone 1 as substrate, the yield of the epoxyketone 2 was always not satisfactory (Scheme 1). In 1999, Crews first reported that epoxidation of 1 with hydrogen peroxide in the presence of diisopropyl ethylamine (DIPEA) and cyanobenzene led to the formation of epoxyketone 2, but a long reaction time of 43 h was needed to acquire 76% yield with a 1.7:1 ( 2a : 2b ) ratio of diastereomers (Scheme 1) [7]. In Proteolix and Onyx patents, epoxidation of 1 was conducted in aqueous N-methyl pyrrolidone, and the using of Ca(OCl)2 as a terminal oxidant, leading to 41% yield of 2 at 0–15 ℃ [8, 9]. Under the same conditions, it afforded a dr ratio of 2:1 in a subsequent patent and the epoxidation of the bis-Boc-protected substrate gave a higher diastereoselectivity with 64% yield [10]. The development of efficient catalytic oxidation method is highly desirable [11, 12], in particular, with earth-abundant metal catalyst (e.g., Mn, Fe) [13].

|
Download:
|
| Fig. 1. Structures of epoxomycin, YU-101 and carfilzomib. | |

|
Download:
|
| Scheme 1. The synthesis of epoxyketone fragment 2 of carfilzomib. | |
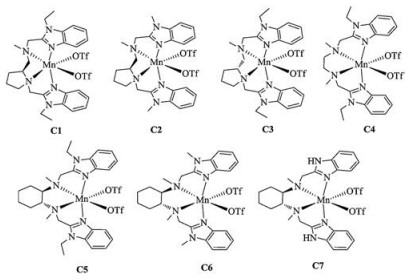
Over the past decades, a great number of chiral manganese complexes with bioinspired tetradentate nitrogen ligands (N4 ligands), as model compounds of nonheme enzymes, have been successfully synthesized and applied to enantioselective epoxidation of olefins [14-20]. Further studies also proved that the addition of organic carboxylic acids, amino acids, even as well as inorganic acids demonstrated important roles in these efficient asymmetric epoxidation methods [21-23]. In our previous study, the manganese catalyst (S-PEB-Mn(OTf)2, Fig. 2, C1) bearing a chiral N4 ligand derived from L-proline and benzimidazole showed high reactivity (up to 99%) and high enantioselectivity (up to 95%) in the epoxidation of α, β-unsaturated ketones. When using the vinyl ketone 1 as substrate, a nearly quantitative yield of epoxyketone 2 was obtained while the ratio of dr ( 2a : 2b ) was only 1:7 [24]. Taking into account the excellent performance of SPEB-Mn(OTf)2 (C1), albeit with epoxyketone 2b as major product, therefore, we anticipate that the manganese complex with opposite configuration or achiral catalyst will give better outcome in this reaction toward the synthesis of valuable intermediate epoxyketone. Herein, a series of bioinspired manganese complexes with N4 supporting ligands, containing benzimidazole motif [25], are carefully investigated in the epoxidation of enone precursor 1 with H2O2 as oxidant in the presence of carboxylic acid (e.g., acetic acid). A gram-scale reaction was also performed under the optimized conditions.

|
Download:
|
| Fig. 2. Structures of the N4 manganese complexes in this work. | |
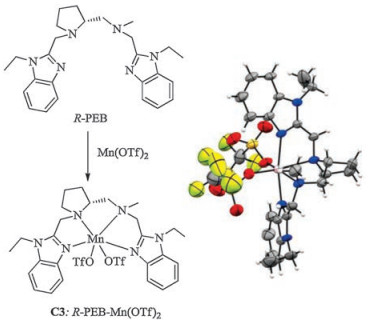
As mentioned above, although the application of C1 produced epoxide in nearly quantitative yield, the proportion of the desired epoxyketone 2a was very low [24]. Clearly, it was caused by the chirality of vinyl ketone 1, and its inherent chirality was mismatched with manganese complex C1. Keep this in mind, a chiral ligand R-PEB derived from D-proline and its manganese complex was easily synthesized based on our previous reported methods (Fig. 3) [24, 25]. Single crystals of C3 (R-PEB-Mn(OTf)2) were obtained by diffusing ether into the CH3CN solution. X-ray crystallographic analysis reveals that the ligand coordinates with manganese center in a cis-α topology as that of manganese complex C3 [24]. Other manganese complexes C1, C2 and C4-C7 were also prepared as previously methods [26, 27].

|
Download:
|
| Fig. 3. The synthesis of R-PEB-Mn(OTf)2 (C3) and crystal structure (CCDC 1870619, atom colors: Mn, pink; C, gray; N, blue; O, red; F, green; S, yellow.) (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article). | |
Initially, we evaluated the activity of C3 in the epoxidation of enone 1. Under the same conditions as our previous reported, in the presence of 5 equiv. of acetic acid in acetonitrile at -20 ℃, manganese complex C3 showed excellent catalytic activity as that of C1 [24]. Epoxyketone 2 was obtained in 97% yield, as expected, and the ration of dr reached to 1:3.1 (Table 1, entry 1). More importantly, the addition of organic carboxylic acid was proved to be effective way to tune the stereocontrol in manganese-catalyzed asymmetric epoxidations through the formation of a carboxylate Mn-oxo intermediate [21-24, 28]. Further, the influence of different organic carboxylic acid was also studied, and the results were summarized in Table 1. When the reaction was performed in the absence of acid, no reaction took place. We could find that the increase of the steric bulk at the α-carbon of the carboxylic acid decrease the reaction activity apparently. For example, the addition of pivalic acid led to a yield less than 8% (Table 1, entry 5) and the addition of 2-ethylbutanoic acid gave no yield of 2 (Table 1, entry 7). As a whole, the organic carboxylic acid played important roles in the epoxidation of 1 to 2, and the best choice is acetic acid. Also, it was found that a higher temperature was adverse for this reaction that the yield of 2 decreased to 77% in the presence of acetic acid at 0 ℃ (Table 1, entry 8).
|
|
Table 1 Catalytic epoxidation of the vinyl ketone 1 to epoxyketone 2 by manganese complex C3a. |

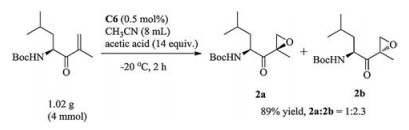
Based on above results, clearly, we can see that the diamine backbone affects the ratio of epoxyketone 2 diastereoselectivity intensively [24, 29]. The catalyst C3 from D-proline let to better results than that of C1. These results further demonstrate that the variation in diastereoselectivity is due to the inherent chirality and steric hindrance of vinyl ketone 1. We next investigated the different manganese complexes C1-C7 (Fig. 2) with various diamines and substituted benzimidazole motifs. The use of achiral manganese complex C4 bearing a 1, 2-ethanediamine led to a lower diastereoselectivity (Table 2, entry 4, 1:5.2). Among the tested catalysts, manganese complex derived from (1R, 2R)-cyclohexane- 1, 2-diamine gave better diastereoselectivity (up to 1:1.6) (Table 2, entries 5–7). On the other hand, the catalytic reactivity and diastereoselectivity were also affected by the steric bulk of N-alkyl group in the benzimidazole of the N4 ligands. We observe that the ratio of dr increases apparently, when changing the N-alkyl group from ethyl to H. The complex C7, without substituent in benzimidazole, gave best dr ratio albeit with low yield under the optimized conditions (Table 2, entry 7, dr = 1:1.6). In order to gain a high yield with a proper dr, 14 equiv of acetic acid was also used and C6 was proved to be the best catalyst by which the yield reached 94% with a dr of 1:2.1. Finally, gram-scale reaction was carried out in order to verify the practical utility of this protocol. As shown in Scheme 2, the epoxyketone 2 was obtained in high isolated (89%) yield and with good diastereoselectivity (1:2.3).
|
|
Table 2 Catalytic epoxidation of the vinyl ketone 1 to epoxyketone 2 by manganese complexes C1-C7a. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}

|
Download:
|
| Scheme 2. The gram-scale synthesis of epoxyketone 2. | |
{kind=link}
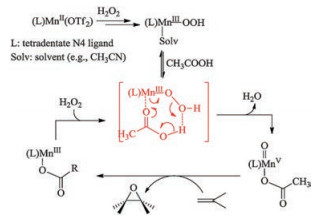
For the manganese-catalyzed epoxidation method, presently, involvement of the high-valent Mn-oxo intermediates has been supported experimentally and theoretically [21-23]. The acetic acid plays a key role during the formation of Mn-oxo, as shown in Scheme 3, acetic acid binding to the metal center facilitates the heterolysis of the O-O bond of the intermediate (MnIII-OOH) [28, 30].

|
Download:
|
| Scheme 3. The proposed mechanism for the manganese-catalyzed epoxidation using H2O2 as the terminal oxidant. | |
{kind=link}
In summary, we report an efficient catalytic epoxidation of the vinyl ketone 1 to the epoxyketone 2, which is the important active component of carfizomib with good ratio of dr, and it could also extend to a gram-scale reaction. Through the study of various organic carboxylic acids as the additives, acetic acid was the best choice for this catalytic system. Further, a series of chiral manganese catalysts with N4 ligands were applied to further study the effects of the chiral ligands on the catalytic reactivity and diastereoselectivity which were proved to be related with the diamine backbone and the steric hindrance. Though the diastereoselectivity of epoxyketone 2 was still challenging, this study provided an alternative method for the synthesis of this key intermediate epoxyketone 2a .
AcknowledgmentsWe acknowledge financial support of this work from the National Natural Science Foundation of China (No. 21473226), Key Research Program of Frontier Sciences, CAS (No. QYZDJ-SSWSLH051) and Natural Science Foundation of Jiangsu Province (Nos. BK20161261 and BK20170420).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi https://doi.org/10.1016/j.cclet.2018.10.023.
| [1] |
K. Redic, J. Pharm. Pharmcol. 65 (2013) 1095-1106. DOI:10.1111/jphp.12072 |
| [2] |
A. Rentsch, D. Landsberg, T. Brodmann, et al., Angew. Chem. Int. Ed. 52 (2013) 5450-5488. DOI:10.1002/anie.201207900 |
| [3] |
K.B. Kim, C.M. Crews, Nat. Prod. Rep. 30 (2013) 600-604. DOI:10.1039/c3np20126k |
| [4] |
D.L. Hughes, Org. Process. Res. Dev. 20 (2016) 2028-2042. DOI:10.1021/acs.oprd.6b00374 |
| [5] |
M.S. Smyth, G.J. Laidig, U.S. Patent, 8207297 B2, 2012.
|
| [6] |
G.J. Laidig, P.A. Radel, M.S. Smyth, U.S. Patent, 0256324 A1, 2005.
|
| [7] |
N. Sin, K.B. Kim, M. Elofsson, et al., Bioorg. Med. Chem. Lett. 9 (1999) 2283-2288. DOI:10.1016/S0960-894X(99)00376-5 |
| [8] |
M.S. Smyth, G.J. Laidig, U.S. Patent, 0094930 A1, 2012.
|
| [9] |
P. Phiasivongsa, L.C. Sehl, W.D. Fuller, G.J. Laidig, U.S. Patent, 8921583 B2, 2014.
|
| [10] |
B. Zheng, Q. Gao, X. Zhang, B. Ding, X. Yang, CN Patent, 104230857 A, 2014.
|
| [11] |
X.L. Ni, J. Liu, Y.Y. Liu, et al., Chin. Chem. Lett. 28 (2017) 1057-1061. DOI:10.1016/j.cclet.2017.01.020 |
| [12] |
S.C. Xu, S.J. Zhu, L.W. Bi, et al., Chin. Chem. Lett. 28 (2017) 575-578. DOI:10.1016/j.cclet.2016.11.020 |
| [13] |
F. Wang, T.Q. Wei, P. Xu, et al., Chin. Chem. Lett. 30 (2019) 379-382. DOI:10.1016/j.cclet.2018.08.006 |
| [14] |
C.X. Miao, C.G. Xia, W. Sun, Sci. Sin. Chim. 44 (2014) 1865-1875. DOI:10.1360/N032014-00199 |
| [15] |
K.P. Bryliakov, E.P. Talsi, Coord. Chem. Rev. 276 (2014) 73-96. DOI:10.1016/j.ccr.2014.06.009 |
| [16] |
A. Murphy, G. Dubois, T.D.P. Stack, J. Am. Chem. Soc. 125 (2003) 5250-5251. DOI:10.1021/ja029962r |
| [17] |
L. Gómez, Garcia-Bosch I., A. Company, et al., Dalton Trans. 47 (2007) 5539-5545. |
| [18] |
M. Wu, B. Wang, S.F. Wang, et al., Org. Lett. 11 (2009) 3622-3625. DOI:10.1021/ol901400m |
| [19] |
R.V. Ottenbacher, K.P. Bryliakov, E.P. Talsi, Adv. Synth. Catal. 353 (2011) 885-889. DOI:10.1002/adsc.v353.6 |
| [20] |
W. Dai, S.S. Shang, B. Chen, et al., J. Org. Chem. 79 (2014) 6688-6694. DOI:10.1021/jo501178k |
| [21] |
O.Y. Lyakin, R.V. Ottenbacher, K.P. Bryliakov, et al., ACS Catal. 2 (2012) 1196-1202. DOI:10.1021/cs300205n |
| [22] |
O. Cussó, Garcia-Bosch I., X. Ribas, et al., J. Am. Chem. Soc. 135 (2013) 14871-14878. DOI:10.1021/ja4078446 |
| [23] |
C.X. Miao, B. Wang, Y. Wang, et al., J. Am. Chem. Soc. 138 (2016) 936-943. DOI:10.1021/jacs.5b11579 |
| [24] |
B. Wang, C.X. Miao, S.F. Wang, et al., Chem. Eur. J. 18 (2012) 6750-6753. DOI:10.1002/chem.201103802 |
| [25] |
J.Y. Du, C.X. Miao, C.G. Xia, et al., Chin. Chem. Lett. 29 (2018) 1869-1871. DOI:10.1016/j.cclet.2018.04.036 |
| [26] |
D.Y. Shen, C.X. Miao, S.F. Wang, et al., Org. Lett. 16 (2014) 1108-1111. DOI:10.1021/ol4037083 |
| [27] |
X.E. Wang, C.X. Miao, S.F. Wang, et al., ChemCatChem 5 (2013) 2489-2494. DOI:10.1002/cctc.201300102 |
| [28] |
J.Y. Du, C.X. Miao, C.G. Xia, et al., ACS Catal. 8 (2018) 4528-4538. DOI:10.1021/acscatal.8b00874 |
| [29] |
D.Y. Shen, C.X. Miao, S.F. Wang, et al., Eur. J. Inorg. Chem. (2014) 5777-5782.
|
| [30] |
H. So, Y.J. Park, K.B. Cho, et al., J. Am. Chem. Soc. 136 (2014) 12229-12232. DOI:10.1021/ja506275q |