 2019, Vol. 30
2019, Vol. 30 


b State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, China;
c School of Pharmacy, Jining Medical University, Rizhao 276826, China;
d Sorbonne Université, Institut Parisien de Chimie Moléculaire, CNRS UMR 8232, 4 place Jussieu, 75005 Paris, France
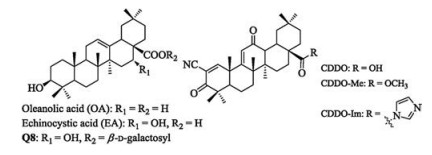
Pentacyclic triterpenes are secondary plant metabolites, widespread in fruit peels, leaves, and stem bark [1], with a few species containing up to 30% of their dry weight [2]. Oleanolic acid (OA, Fig. 1), an oleanane-type pentacyclic triterpene, has been isolated from more than 1, 600 plant species, including many dietary and medicinal plants [3]. Numerous studies indicate natural OA and its saponins exhibit a wide range of pharmacological activities, such as antiviral, antitumor, anti-inflammation, and anti-microbial activities [4]. Due to its inherent bioactivities, availability and low cost, OA has been used as a starting molecule for the synthesis of oleanane triterpenes. A series of novel synthetic oleanane triterpenes have been prepared by chemical modifications of OA at three sites, including C-3 hydroxyl, △12-13 double bond and C-28 carboxylic acid [5]. Several synthetic derivative of OA, 2-cyano-3, 12-dioxoolean-1, 9-dien-28-oic acid (CDDO) and its C-28 methyl ester (CDDO-Me) and C-28 imidazole (CDDO-Im) derivatives, have been tested in clinical trials [6]. Recently, we found that echinocystic acid (EA), an analog of OA with an extra hydroxyl group at C-16, displays substantial inhibitory activity on hepatitis C virus (HCV) entry with one derivative Q8 (Fig. 1) showing EC50 at nanomolar level [7]. Despite their promising bioactivity, pentacyclic triterpenes are bulky, nonpolar, and poorly soluble in water, which are major drawbacks for various applications, in particular, the development of drugs. For example, the solubility of OA in water is only about 0.05 mg/mL [8]. Hence, the synthesis of more water-soluble triterpene derivatives is needed. Some water-soluble derivatives have been synthesized by introducing polar sugar moieties at C-3 and/or C-28 [9].

|
Download:
|
| Fig. 1. The chemical structures of OA and its derivatives. | |
{kind=link}
β-Cyclodextrin (CD) is a macrocyclic oligomers of D-glucose with the secondary C-2 and C-3 hydroxyl groups on the secondary face and the primary C-6 hydroxyl group on the first face. This unique structure enables β-CD to possess high aqueous solubility. Due to the ability to form inclusion complex with a large range of hydrophobic drug molecules, the physicochemical properties of drugs are significantly modified after forming complexes with β-CD, such as the increased stability and water solubility, enhanced dissolution rate and bioavailability [10]. Several CD inclusion complexes with triterpenes have been studied [11]. Usually, the drugs are released quickly from the inclusion complex under physiological conditions. Alternatively, direct covalent linkage with CD has been suggested to improve the physicochemical properties of triterpines, which has been widely used for other water insoluble bioactive molecules, such as fluorouracil (5-FU) and folic acid [12].
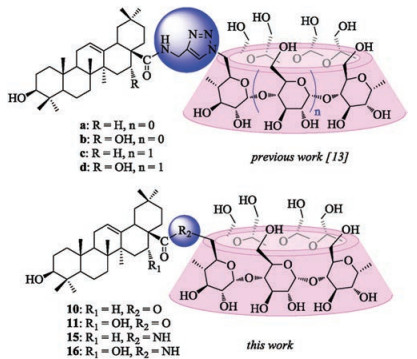
In our previous studies, a series of water-soluble triazolebridged α- and β-CD-pentacyclic triterpene conjugates have been synthesized via "click chemistry" (Fig. 2) [13]. As a continuation towards the elucidation of the relationship between the structure and anti-HCV entry activity of triterpene derivatives, we have synthesized conjugates a-d in which the triazolyl linker was replaced by ester or amide bond. Herein, we describe a simple method to prepare those β-CD derivatives and their lipophilicity (AlogP) along with their cytotoxicity to three human cancer cell lines.

|
Download:
|
| Fig. 2. Structure of novel 1:1 CD-pentacyclic triterpene conjugates. (Top: linked by triazol bond [13], bottom: linked by ester or amide bond). | |
{kind=link}
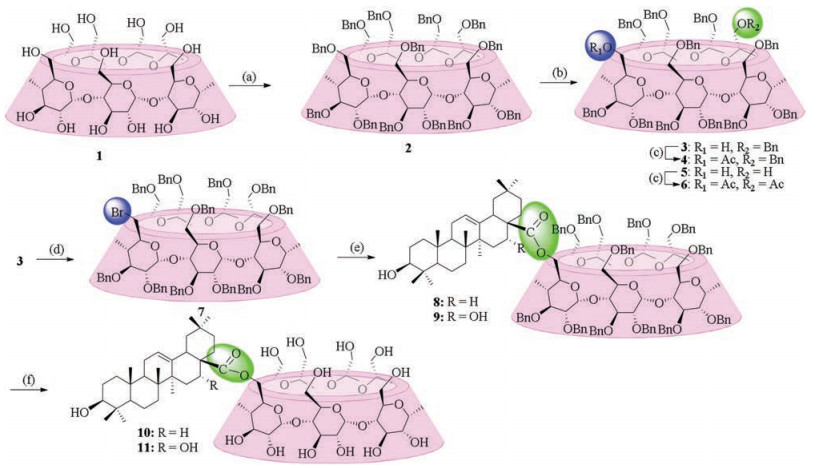
As shown in Scheme 1, conjugation of β-CD with two oleananetype triterpenes OA and EA via ester bond started from 6A-hydroxyper-O-benzyl-β-CD (3), a regioselective mono-debenzylated intermediate. 3 was prepared from the per-O-benzylated β-CD using diisobutylaluminium hydride (DIBAL-H) (30 equiv.) as a chemical "scalpel" as first introduced by P. Sinay group [14]. In addition to € the mono-debenzylated β-CD (3), the di-de-O-benzylated product 5 was also obtained in 20% yield after chromatography separation. The structures of 3 and 5 were further confirmed by their acetylated derivatives 4 and 6, respectively. Initial attempts using classical methods to conjugate triterpenes OA and EA with 3 directly via ester bond yielded no desired product. Therefore, the compound 3 was converted to its bromide derivative 7 using the commercially available carbon tetrabromide (CBr4) and triphenylphosphine (PPh3) in 85% yield. It was characterized using 1H and 13C NMR, which gave the distinctive bromine methyl signals at 3.55 and 3.80 ppm (2 × H6A) and 34.59 ppm (C6A), respectively.

|
Download:
|
| Scheme 1. Synthesis of β-CD-oleanane-type triterpene conjugates 10 and 11 via ester bond. Reagents and conditions: (a) BnBr, NaH, DMF, 86%; (b) DIBAL-H (30 equiv.), toluene, 50 °C, 2h, 42.1% for compound 3 and 20.0% for compound 5; (c) Ac2O, pyridine, DMAP, 81%-85%; (d) CBr4, PPh3, CH2Cl2, 85%; (e) EA or OA, K2CO3, DMF, 62-82%; (f) Pd/C, H2, CH3OH, 78%-91%. | |
{kind=link}
Conjugation of the key intermediate 7 with triterpenes EA and OA in the presence of K2CO3 provided compound 8 or 9 smoothly in 88%-92% yield, respectively. Fig. S1 in Supporting information presents the 1H and 13C NMR spectra of compound 9. The triplet at 5.33 ppm (J = 3.0 Hz) refers to 1H, which, according to its 1H-1H and 1 H-13C correlation spectra, should be assigned to H12. The highfield doublets of doublet at 3.16 ppm (J = 4.4 and 11.1 Hz) and 3.05 ppm (J = 3.5 and 14.5 Hz), each referring to 1H, are assigned to H3 and H18, respectively. The ESI-HRMS mass spectrum of compound 9 recorded in the positive mode shows clearly an [M/2+NH4]+ ion at m/z 1712.8676 (calcd. isotopic mass for 12C1061H12214N116O19: 1712.8606), indicating that it was indeed connected via an ester bond. Similar patterns were also observed in 1H NMR and 13C NMR spectra of compound 8 of this series. Finally, de-O-benzylation of 8 or 9 was accomplished by hydrogenolysis to yield 10 or 11 in high yields (78%-91%).
A parallel experiment was carried out to conjugate pentacyclic triterpene with β-CD moiety through an amide bond rather than an ester bond. Such alteration might enhance the stability of pentacyclic triterpene-β-CD conjugates. As shown in Scheme 2, selective monotosylation of β-CD (1) according to the procedure described by Vizitiu et al. [15] with minor modifications afforded compound 12, which was used without further purification for the nucleophilic substitution with NaN3 in DMF to provide the monoazide substituted derivative 13 in 8% yield over two steps. Reduction of the azide group of 13 with triphenylphospine followed by hydrolysis gave the key intermediate 6A-amino-6Adeoxy-β-CD 14. Finally, conjugation of 14 with triterpenes OA and EA in the presence of N-ethyl-N'-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC°HCl) afforded compounds 15 and 16 in moderate yields (43%-47%). Structural elucidation of the new derivatives was also done using 1D-NMR (1H and 13C), 2D-NMR and MALDI-TOF-MS techniques (Supporting information).

|
Download:
|
| Scheme 2. Synthesis of β-CD-pentacyclic triterpene conjugates 15 and 16 via amide bond. Reagents and conditions: (a) H2O, NaOH, TsCl, 9.4%; (b) NaNaN3, DMF, 80 °C, 91%; (c) Ph3P, DMF, NH3 (aq), 63%; (d) EDC, HOBt, DMF, 43%-47%. | |
{kind=link}
The 1-octanol/water partition coefficient (logP) is a well-known measure of molecular lipophilicity. In a recent study, a negative correlation was found between the llogP value of terpenes and their bioaccessibility (r = - 0.77, P < 0.001) [16]. Therefore, decrease in the lipophilicity of pentacyclic triterpenes may increase the bioaccessibility. In this study, the calculated AllogP values based on Ghose and Crippen's method were determined using Pipeline Pilot software, Vers. 7.5 (Accelrys Corporation, San Diego, USA) [17]. We found that the targeting conjugates 10/11 and 15/16 showed increased hydrophilicity comparing with their parent compounds due to the introduction of β-CD (Table S1 in Supporting information). Compared with their analogs b and d [13a], the ALlogP of conjugates 10 and 11 increased by 0.66 log units (triazol linker vs. ester linker) while the ALlogP of conjugates 15 and 16 remained almost the same (trizolyl linker vs. amide linker). In addition, due to the 16-hydroxyl group of EA, the ALlogP of conjugates 11 and 16 decreased by 1.1 log units (10 vs. 11 and 15 vs. 16), which means that their solubility in water are increased by ~ 12.5–fold.
Oleanane-type triterpenes are widely found in the plant kingdom as free acids or triterpene saponins linked with one or more sugar chains. Some natural pentacyclic triterpenes are shown weak cytotoxicity [18]. In this study, the cytotoxicity of the four conjugates (10, 11, 15 and 16) against human promyelocytic leukemia (HL-60), human cervical cancer (Hela), human lung cancer (A549), and human liver cancer (Bel-7402) cell lines was examined by the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay at concentrations of 0.1-10 mmol/L [19]. Except conjugates 10, 11 and 15 showed weak cytotoxicity to Hela cell at concentration of 1 mmol/L and 10 mmol/L (~ 23%-26%), no significant cytotoxicity was observed at concentrations up to 10 mmol/L for other three cancer cell (Table S2 in Supporting information). These results indicated that the synthesized β-CDpentacyclic triterpene derivatives had no significant toxic effect to cells in vitro.
In summary, starting from natural β-CD, four water-soluble oleanane-type triterpene-β-CD conjugates 10/11 and 15/16 were synthesized via ester and amide linkage by a facile method. The new products were unambiguously characterized by 1H NMR, 13C NMR and HRMS or MALDI-TOF-MS. Compared with their parent compounds, they showed higher hydrophobicity (AllogP) with no significant cytotoxicity in vitro. This study supports that these conjugates are potential candidates for further evaluation as antiHCV entry agents.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 81573269, 21572015, 21877007, 91753202 and 21702007) and the open funding of the State Key Laboratory of Phytochemistry and Plant Resources in West China. The authors gratefully acknowledge Dr. Hongwei Jin at State Key Laboratory of Natural and Biomimetic Drugs, Peking University for carrying out the ALlogP determinations and Dr. Lijun Zhong at the Centre of Medical and Health Analysis, Peking University for MALDI-TOF experiments.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2018.10.009.
| [1] |
J.T. James, I.A. Dubery, Molecules 14 (2009) 3922-3941. DOI:10.3390/molecules14103922 |
| [2] |
S. Alakurtti, T. Makela, S. Koskimies, Yli-Kauhaluoma J., Eur. J. Pharm. Sci. 29 (2006) 1-13. DOI:10.1016/j.ejps.2006.04.006 |
| [3] |
J. Pollier, A. Goossens, Phytochemistry 77 (2012) 10-15. DOI:10.1016/j.phytochem.2011.12.022 |
| [4] |
Rodriguez-Rodriguez R., Curr. Med. Chem. 22 (2015) 1414-1425. DOI:10.2174/0929867322666141212122921 |
| [5] |
M.B. Sporn, K.T. Liby, M.M. Yore, et al., J. Nat. Prod. 74 (2011) 537-545. DOI:10.1021/np100826q |
| [6] |
(a) D.S.Hong, R.Kurzrock, J.G.Supko, etal., Clin.CancerRes.18 (2012)3396-3406; (b)M.K. Shanmugam, X.Y. Dai, A.P. Kumar, et al., Cancer Lett. 346 (2014) 206-216. |
| [7] |
F. Yu, Q. Wang, Z. Zhang, et al., J. Med. Chem. 56 (2013) 4300-4319. DOI:10.1021/jm301910a |
| [8] |
L.X. Liu, X.C. Wang, J. Chem. Eng. Data 52 (2007) 2527-2528. DOI:10.1021/je700312r |
| [9] |
K. Xu, F.H. Chu, G.L. Li, et al., Pharmazie 69 (2014) 483-495. |
| [10] |
(a) B. Gidwani, A. Vyas, Biomed. Res. Int. 2015 (2015) 198268; (b) S.V. Kurkov, T. Loftsson, Int. J. Pharm. 453 (2013) 167-180. |
| [11] |
(a) H.M. Wang, C.M. Soica, G. Wenz, Nat. Prod. Commun. 7 (2012) 289-291; (b) A. Hertrampf, C. Grundemann, S. Jager, et al., Planta Med 78 (2012) 881-889; (c) C. Soica, C. Oprean, F. Borcan, et al., Molecules 19 (2014) 4924-4940. |
| [12] |
(a) K. Udo, K. Hokonohara, K. Motoyama, et al., Int. J. Pharm. 388 (2010) 95-100; (b) R. Onodera, K. Motoyama, A. Okamatsu, T. Higashi, H. Arima, Sci. Rep. 3 (2013) 1103. |
| [13] |
(a) S. Xiao, Q. Wang, L. Si, et al., ChemMedChem. 9 (2014) 1060-1070; (b) S. Xiao, Q. Wang, L. Si, et al., Eur. J. Med. Chem. 124 (2016) 1-9. |
| [14] |
A.J. Pearce, P. Sinay, Angew. Chem. Int. Edit. 39 (2000) 3610-3612. |
| [15] |
D. Vizitiu, C.S. Walkinshaw, B.I. Gorin, G.R.J. Thatcher, J. Org. Chem. 62 (1997) 8760-8766. DOI:10.1021/jo9711549 |
| [16] |
D. Martin, Navarro del Hierro J., Villanueva Bermejo D., et al., J. Agr. Food Chem. 64 (2016) 8828-8837. DOI:10.1021/acs.jafc.6b04313 |
| [17] |
(a) Q. Shi, T.M. Kaiser, Z.W. Dentmon, et al., ACS Med. Chem. Lett. 6 (2015) 518-522; (b) F. Lovering, J. Bikker, C. Humblet, J. Med. Chem. 52 (2009) 6752-6756. |
| [18] |
(a) P. Dzubak, M. Hajduch, D. Vydra, et al., Nat. Prod. Rep. 23 (2006) 394-411; (b) K.T. Lee, J. Choi, W.T. Jung, et al., J. Agr. Food Chem. 50 (2002) 4190-4193. |
| [19] |
T. Mosmann, J. Immunol. Methods 65 (1983) 55-63. DOI:10.1016/0022-1759(83)90303-4 |