 2019, Vol. 30
2019, Vol. 30 


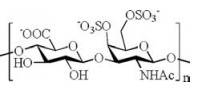
Chondroitin sulfates are classified into certain categories based on the sulfation pattern of the repeating disaccharide moieties. As one subtype of CS, chondroitin sulfate E (CS-E), possessing sulfate groups at the 4 and 6 positions of the GalNAc residue (Fig. 1), has particular biological properties [1-10], such as the ability to inhibit neuronal cell adhesion [6] and interact with various heparinbinding growth factors [9].

|
Download:
|
| Fig. 1. Structure of the basic repeating unit of chondroitin sulfate E. | |
{kind=link}
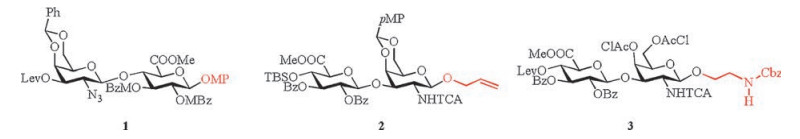
Significant amounts of structurally well-defined oligosaccharide sequences are needed to better understand the pharmacological functions of CS-E at the molecular level. However, it is not easy to obtain structurally defined CS-E oligosaccharides from natural sources. To fulfill this need, several chemical syntheses of CS-E oligosaccharides equipped with different anomeric groups have been published [11-21]. In all of these approaches, anomeric groups were first introduced in the disaccharide unit (Fig. 2) [12-18], followed by a lengthening of the chain while maintaining the same group at the anomeric center. No efficient general method has been reported to prepare size-defined oligosaccharides with different groups at the anomeric center. An expeditious and stereocontrolled method to prepare oligosaccharide precursors for further preparation of chondroitin sulfate E is urgently needed and motivates this study. Herein, we report a procedure for preparing several CS-E oligosaccharide precursors with different anomeric groups that can be easily transferred into various CS-E oligosaccharides for further structure-activity relationship investigations.

|
Download:
|
| Fig. 2. Structures of key disaccharide units used in the total synthesis of CS-E oligosaccharides. Abbreviations: Lev, MeCO(CH2)2CO–; MBz, p-MeC6H4CO–; MP, p-MeOC6H4–; TCA, Cl3CCO–; Bz, C6H4CO–; Cbz, C6H4CH2OCO–. | |
{kind=link}
Although several syntheses of CS-E oligosaccharides have been reported [12-20], the traditional synthetic strategy of first introducing anomeric groups followed by elongation of the oligosaccharides via 2 + 2 or 2 + 4 glycosylation coupling was deemed inefficient to synthesize a set of oligosaccharides equipped with different anomeric groups. Therefore, a more efficient synthetic strategy was required to obtain more oligosaccharides for further preparation of CS-E. A new synthetic strategy was envisioned to reduce the number of chemical transformations. The disaccharide unit was first lengthened to a tetra- or hexasaccharide while maintaining the same group at the anomeric center. Removal of the anomeric group was followed by imidoylation to produce a common imidate precursor. Starting from the common intermediate, a short route to gain access to a set of glycosides was planned via trichloroacetimidate glycosylation.
Based on these ideas, we synthesized several CS-E tetrasaccharide precursors (Scheme 1). The disaccharide of thioglycoside 4 was chosen as the starting material [21], which was either transformed into acceptor 6 through selective delevulinoylation with hydrazine hydrate, or into donor 5 through the hydrolysis of the 4-methylphenylthiol group with N-iodosuccinimide/trifluoroacetic acid (NIS/TFA) followed by trichloroacetimidoylation. Then glycosylation coupling of imidate 5 with acceptor 6 under the catalysis of trimethylsilyl triflate (TMSOTf) gave the tetrasaccharide 7 in 80% yield. The common precursor 8 was a key intermediate and was obtained via a similar procedure as compound 5. Glycosylation coupling of imidate 8 with selective alcohols under the catalysisof TMSOTf afforded exclusively the 1, 2- trans-linked glycosides 9–13 in excellent yields (74%–92%). The structures of all the compounds were confirmed by 1H NMR, 13C NMR and HR-MS. The detailed synthetic procedure and related spectral data can be found in Supporting information.

|
Download:
|
| Scheme 1. Synthesis of chondroitin sulfate E tetrasaccharide precursors. Reagents and conditions: (a) NIS, TFA, DCM/H2O, r.t., 5h; (b) CCl3CN, DBU, DCM, r.t., 2 h, 81% for two steps; (c) NH2NH2°H2O, Py, AcOH, r.t., 89%; (d) TMSOTf, DCM, 4Å-MS, -60 ℃, 0.5h, 80%; (e) NIS, TFA, DCM, 0 ℃; (f) CCl3CN, DBU, 0 ℃, DCM, 72% for twosteps; (g) TMSOTf, DCM, 4Å-MS, -20 ℃ to 0 ℃. Abbreviations: PMB, p-MeOC6H4CH2–; Tol, p-MeC6H4–; TMS, (CH3)3Si–. | |
{kind=link}
To highlight the advantage of the new synthetic approach, the tetrasaccharide precursors 9 and 10 were synthesized via the traditional route (Scheme 2). Anomeric groups were introduced in disaccharide units, which can be easily transformed into acceptor 14 or 15 after selective delevulinoylation. The detailed synthesis of compounds 14 and 15 can be found in Supporting information. Glycosylation coupling of imidate 5 with acceptor 14 or 15 under the catalysis of TMSOTf gave tetrasaccharides 9 and 10. Meanwhile, a temperature screening (-60 ℃ to r.t.) of the glycosylation coupling indicated that 0 ℃ was optimal, with 9 and 10 obtained in 64% and 35% yields, respectively.

|
Download:
|
| Scheme 2. Synthesis of tetrasaccharide precursors 9 and 10 by the traditional route. | |
{kind=link}
The new synthetic approach offers successful improvement versus the traditional route. The disaccharide was first lengthened to a tetrasaccharide and then transformed into the imidate. The divergent approach for the construction of glycosides 9–13 from a single precursor was more efficient, and all the key glycosylation steps were in high yields.
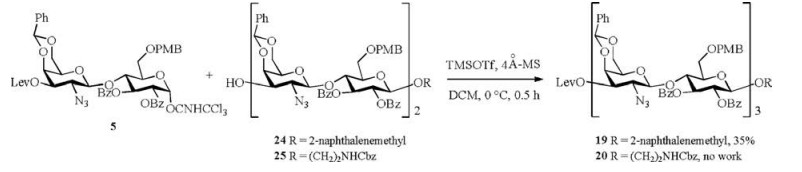
With tetrasaccharide 7 in hand, the synthesis of several hexasaccharide precursors was next undertaken via this new synthetic approach (Scheme 3). Selective removal of the levulinoyl group in compound 7 with the hydrazine hydrate gave acceptor 16 in 88% yield. Glycosylation of imidate 5 with acceptor 16, as described for the preparation of 7, afforded hexasaccharide 17 in 71% yield. As described for the preparation of 8, the key intermediate imidate 18 was obtained in 57% yield over two steps. Hexasaccharide imidate 18 was transformed into the corresponding glycosides 19-23 in the presence of TMSOTf (20% based on 18) in dichloromethane at 0 ℃ or below. All glycosylation steps resulted in good yields (70%-87%). The structures of all the compounds were confirmed by 1H NMR, 13C NMR and HR-MS. The detailed synthetic procedure and related spectral data can be found in Supporting information.

|
Download:
|
| Scheme 3. Synthesis of chondroitin sulfate E hexasaccharide precursors. Reagents and conditions: (a) NH2NH2·H2O, Py, AcOH, r.t., 88%; (b) donor 5, TMSOTf, DCM, 4Å-MS, -60 ℃, 0.5 h, 71%; (c) NIS, TFA, DCM, 0 ℃, 5 h; (d) CCl3CN, DBU, 0 ℃, DCM, 57% for two steps; (e) TMSOTf, DCM, 4Å-MS, -20 ℃ to 0 ℃. | |
{kind=link}
Two anomeric groups were introduced to hexasaccharide following the same traditional synthetic approach as above to compare synthetic approaches (Scheme 4). Glycosylation coupling of imidate 5 with the acceptor compound 24 gave the hexasaccharide derivative 19 in 35% yield. The synthesis of compounds 24 and 25 was deposited in Supporting information. Coupling imidate 5 with compound 25, however, did not result in the expected corresponding hexasaccharide derivative 20, even under vigorous reaction conditions.

|
Download:
|
| Scheme 4. Synthesis of hexasaccharide precursors 19 and 20 by the traditional route. | |
{kind=link}
This new strategy was deemed more suitable to lengthen chain to hexasaccharide anomeric species versus the traditional synthetic approach in excellent yields, especially 20 which was equipped with a 2-benzyloxycarbonylaminoethyl group at the anomeric center.
Comparison of the two synthetic approaches revealed that the new approach has advantages over the traditional route. For one thing, the new synthetic approach was an efficient, reliable, stereocontrolled and highly divergent approach for the preparation of a set of CS-E oligosaccharide precursors with different anomeric groups. A common imidate was easily prepared and then used as a precursor for a highly divergent chemical synthesis of all anomeric variants. The glycosylation steps were in high yields. Moreover, the new synthetic approach was suitable to introduce the different anomeric groups we designed, and the overall yields were significantly improved, especially for installing the 2- benzyloxycarbonylaminoethyl group to hexasaccharide. Most notably, the new strategy reduces the number of steps and avoids having to repeat the same sequence of reactions starting from disaccharide units.
In summary, a facile and efficient approach has been developed for the construction of CS-E oligosaccharide precursors. In this approach, a disaccharide unit was elongated to tetra- and hexasaccharides, followed by the introduction of anomeric groups via glycosylation couplings. Several CS-E tetra- and hexasaccharide precursors were prepared. It is hoped that these precursors and their transformations into CS-E oligosaccharides will help in understanding the intriguing biological functions and the structure–activity relationships of these natural products. In addition, the strategy employed here can be extended for the preparation of larger chondroitin oligomers and similar members of the glycosaminoglycan family.
AcknowledgmentsThis work was supported by the CAMS Innovation Fund for Medical Sciences (Nos. CIFMS 2016-I2M-3-009 and CIFMS 2017- I2M-3-011). And we are grateful to the Department of Instrumental Analysis of our institute for performing the NMR and mass spectroscopy measurements.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.06.012.
| [1] |
K. Sugahara, T. Mikami, T. Uyama, et al., Curr. Opin. Struct. Biol. 13 (2003) 612-620. DOI:10.1016/j.sbi.2003.09.011 |
| [2] |
K. Kadomatsu, T. Muramatsu, Cancer Lett. 204 (2004) 127-143. DOI:10.1016/S0304-3835(03)00450-6 |
| [3] |
C.I. Gama, Hsieh-Wilson L.C., Curr. Opin. Chem. Biol. 9 (2005) 609-619. DOI:10.1016/j.cbpa.2005.10.003 |
| [4] |
L. Fuchuan, D.N. Chilkunda, S. Kazuyuki, J. Biol. Chem. 285 (2010) 27673-27685. DOI:10.1074/jbc.M110.118703 |
| [5] |
C. Ueoka, N. Kaneda, I. Okazaki, S. Nadanaka, et al., J. Biol. Chem. 275 (2000) 37407-37413. DOI:10.1074/jbc.M002538200 |
| [6] |
K. Takagaki, H. Munakata, I. Kakizaki, et al., J. Biol. Chem. 277 (2002) 8882-8889. DOI:10.1074/jbc.M106479200 |
| [7] |
H. Kawashima, K. Atarashi, M. Hirose, et al., J. Biol. Chem. 277 (2002) 12921-12930. DOI:10.1074/jbc.M200396200 |
| [8] |
S. Deepa, Y. Umehara, S. Higashiyama, N. Itoh, K. Sugahara, J. Biol. Chem. 277 (2002) 43707-43716. DOI:10.1074/jbc.M207105200 |
| [9] |
L. Fuchuan, D.N. Chilkunda, S. Kazuyuki, J. Biol. Chem. 285 (2010) 27673-27685. DOI:10.1074/jbc.M110.118703 |
| [10] |
V. Aude, L.B. Chrystel, Jean-Claude J., Tetrahedron Lett. 51 (2010) 1867-1869. DOI:10.1016/j.tetlet.2010.02.005 |
| [11] |
J.I. Tamura, Tsutsumishita-Nakai N., Y. Nakao, et al., Bioorg. Med. Chem. Lett. 22 (2012) 1371-1374. DOI:10.1016/j.bmcl.2011.12.054 |
| [12] |
J.I. Tamura, M. Tokuyoshi, Biosci. Biotechnol. Biochem. 68 (2004) 2436-2443. DOI:10.1271/bbb.68.2436 |
| [13] |
J.I. Tamura, N. Yuka, T. Kayo, Y. Manami, Carbohydr. Res. 343 (2008) 39-47. DOI:10.1016/j.carres.2007.09.009 |
| [14] |
S.E. Tully, R. Mabon, Hsieh-Wilson L.C., et al., J. Am. Chem. Soc. 126 (2004) 7736-7737. DOI:10.1021/ja0484045 |
| [15] |
R. Manish, G.I. Cristal, Hsieh-Wilson L.C., et al., J. Am. Chem. Soc. 130 (2008) 2959-2961. DOI:10.1021/ja709993p |
| [16] |
A. Katia, L.B. Chrystel, J.C. Jacquinet, Carbohydr. Res. 353 (2012) 33-48. DOI:10.1016/j.carres.2012.03.039 |
| [17] |
J.C. Jacquinet, L.B. Chrystel, Carbohydr. Res. 402 (2015) 35-43. DOI:10.1016/j.carres.2014.09.007 |
| [18] |
J.C. Jacquinet, em Ch, Eur. J. 15 (2009) 9579-9595. DOI:10.1002/chem.v15:37 |
| [19] |
J.C. Jacquinet, L.B. Chrystel, V. Aude, Chem. Eur. J. 15 (2009) 9561-9578. DOI:10.1002/chem.v15:37 |
| [20] |
S. Yang, P. A.-Wang, G.Y. Zhang, et al., Tetrahedron. 72 (2016) 5659-5670. DOI:10.1016/j.tet.2016.07.042 |
| [21] |
S. Yang, Q. Liu, G.Y. Zhang, et al., J. Org. Chem. 83 (2018) 5897-5908. DOI:10.1021/acs.joc.8b00157 |