 2019, Vol. 30
2019, Vol. 30 


b College of Physics, Optoelectronics and Energy & Collaborative Innovation Center of Suzhou Nano Science and Technology, Soochow University, Suzhou 215006, China
Highly efficient and green energy conversion and storage devices, such as zinc-air batteries, fuel cells, and water splitting devices, are attracting tremendous attention due to the global energy and environment challenges [1-5]. The oxygen reduction reaction (ORR) is the vital reaction in zinc-air batteries [6-9] and fuel cells [10-12], while the hydrogen evolution reaction (HER) is the key reaction in water splitting devices [13-15]. The Pt/C catalysts are the state-of-the-art catalysts for ORR and HER, but their disadvantages, including crossover deactivation, poor durability and high cost, hampered their large-scale application [1, 4, 6, 16]. Consequently, the highly efficient, stable and cheap electrocatalysts for ORR and HER are urgently needed. Nitrogen doped porous carbons are considered as the promising catalysts for both ORR and HER because of hierarchical porous structure, high specific surface area and nitrogen-containing active sites [1, 17]. On the other hand, the embedment of transition metal-atoms in the nitrogen doped carbons could further enhance the electrochemical activity for ORR and HER [16]. Therefore, the transition metal and nitrogen codoped carbons (M-N-C) could be a quite promising electrocatalyst for ORR and HER. A large variety of nitrogencontaining substances have been employed as the precursors of MN-C. These precursors mainly included natural macromolecules, synthetic polymers and organic molecules, such as chitosan [18], polypyrrole [19], polydopamine [20], polyaniline [21, 22], polyacrylonitrile [23], 2-methylimidazole [1, 24-26], corrole [27, 28], bipyridine [29] and cyanamide [11]. The kind of nitrogencontaining precursors greatly affected the performances of the M-N-C electrocatalysts [30-32].
Some nitrogen-containing synthetic polymers such as poly(4- vinylpyridine) (P4VP), poly(ethyleneimine) (PEI), and polyvinylpyrrolidone (PVP) decomposed completely and showed no residue at high temperature even in inert gases [33-35], so they are generally regarded as "noncarbonizable" polymer. Interestingly, they could form coordination network with metal ions and then transform to porous carbon electrocatalysts by pyrolysis [33, 36-38]. For example, Shi and co-workers mixed PEI with ferrous sulfate to form coordination polymers and then pyrolyzed them to prepare the porous carbons for ORR catalyst [36]. Li et al. used PVP as precursor and ferrous acetate as coordination agent to fabricate the porous carbon electrocatalyst for water splitting [38]. However, these "noncarbonizable" polymers have not been utilized to fabricate the M-N-C bifunctional catalysts for ORR and HER.
In this work, P4VP and cobalt chloride hexahydrate were mixed to form coordination polymer and then pyrolyzed using SiO2 nanoparticles as hard template.Theas-made cobalt and nitrogen codoped carbons (Co-N-C) displayed excellent bifunctional electrocatalystic performances for both ORR and HER in KOH medium. In particular, their stability of catalysis for ORR and HER distinctly outperformed commercial Pt/C catalyst and other bifunctional M-N-C catalysts in alkaline solution [1, 23, 39]. To our knowledge, these Co-N-C materials are the first M-N-C bifunctional electrocatalyst derived from the "noncarbonizable" polymers for ORR and HER.
P4VP degraded completely and showed no residue at ~500 ℃ under nitrogen atmosphere (Fig. S1 in Supporting information), similartoPEIandPVP, so it was not employed as a precursor of carbon material until four years ago [36]. In this work, the mixtures of P4VP, CoCl2-6H2O and SiO2 nanoparticles generated the carbon materials after drying, pyrolysis, HF etching, and water washing because the coordination effect between P4VP and metal ion (Co2+) facilitated P4VP to form carbon materials at high temperature (details in Supporting information). The preparation conditions and product codes are compiled in Table S1 (Supporting information). The "x" of the product codes (Co-N-C-x) stood for the pyrolysis temperature. The product of Co-N-C-800 exhibited high thermal stability, the weight loss of which was only 6% at 800 ℃ (Fig. S1). Similar "noncarbonizable" polymers were also transformed to carbon materials at high temperature by forming coordination polymers recently [36]. Herein P4VP was used as both carbon and nitrogen sources, CoCl2·6H2O was employed as complexant and cobalt source, while the SiO2nanoparticles with adiameter of ~10 nm were utilized as hard template. After HF etching, the SiO2 nanoparticles were removed from the product and thus generated lots of mesopores in the resulting materials (Co-N-C).
The chemical composition and structure of the as-made products were characterized by XRD, XPS, ICP, Raman, SEM, and TEM. The XRD patterns are exhibited in Fig. S2a (Supporting information). As shown, Co-N-C-700, -800 and -900 exhibited a very broad diffuse peaks centered at 30.0°, which corresponded to the (002) plane of carbon material [36]. Meanwhile, this peak (002) possessed two diffuse shoulder peaks, one centered at 41.7° originated from the (100) plane of carbon material [29, 40], while the other shoulder peak was at ~17.9°, indicating an enhanced interlayer spacing because of nitrogen doping [41-43]. The XRD diffraction pattern of Co-N-C-800 (before HF etching) in Fig. S2b (Supporting information) could match the standard XRD pattern of Co (Card No. 06-0727 and 15-0806), revealing the existence of Co crystal. After HF etching, all diffraction peaks of the crystalline Co disappeared, which meant that the HF etching process removed the surface cobalt particles, except for the SiO2 nanoparticle template. As expected, the mixture of the precursors displayed all the characteristic peaks of raw CoCl2-6H2O but exhibited no other diffraction peaks (Fig. S2c in Supporting information).
The surface chemical composition of Co-N-C catalysts was measured by XPS. Their XPS survey spectra are presented in Fig. 1. These XPS survey spectra illustrated that the N and Co elements were doped to the resulting carbon materials. As expected, the O element was also doped in the products like many previous reports, which originated from the SiO2 nanoparticles [6, 44-47]. As indicated in Table S2 (Supporting information), Co-N-C-700, -800, and -900 show a nitrogen content of 5.43 at%, 4.27 at%, and 3.79 at%, respectively. Co-N-C-900 exhibited the lowest nitrogen content among them, attributing to the decomposition of the Ncontaining structure at high pyrolysis temperature [48]. In Fig. 1b, the N 1s curves of Co-N-C catalysts are divided into four peaks, corresponding to the pyridinic N (398.4 eV, N1), pyrrolic N (399.6 eV, N2), graphitic N (401.0 eV, N3), and oxidized N (403.0 eV, N4), respectively [41, 49-52]. Because the binding energy of nitrogen bound to metal atom (M-N) was very close to that of pyridinc N, the content of pyridinc N included a contribution from M-N [44, 53]. It is generally believed that the pyridinc N and graphitic N played a positive effect on ORR and HER, whereas the pyrrolic N had a negative effect [1, 44, 53-56]. In Fig. 1c, the ratios of pyridinc and graphitic N to pyrrolic N in Co-N-C-700, -800 and -900 are 5.26, 8.76 and 5.53, respectively, which could reflected the electrocatalytic performance to some extent [57]. Compared with Co-N-C-700 and -900, Co-N-C-800 possessed the highest ratio of pyridinc and graphitic N to pyrrolic N, suggesting that the catalytic activity of Co-N-C-800 might be better than other two ones. Besides, in Fig. 1d, the XPS peaks of Co 2p3/2 and Co 2p1/2 could be identified at a binding energy of 780.5 and 796.3 eV, respectively, though their peak intensity was very weak. At the same time, the peak of Co 2p3/2 of Co-N-C-800 could be deconvoluted into two peaks, corresponding to the nitrogen-coordinated cobalt (779.8 eV) and oxygen-coordinated cobalt (781.5 eV), respectively (Fig. S3a in Supporting information) [30]. Because the intensity of the XPS peaks of Co element was very weak, the Co content derived from XPS peaks was not very accurate. In order to accurately determine the Co quantity of Co-N-C, the ICP measurements were performed. The ICP results are compiled in Table S2. As shown, CoN-C-800 showed a Co content of 0.49 wt%, close to (0.48 wt%) that of Co-N-C-700, but higher than that (0.33 wt%) of Co-N-C-900.

|
Download:
|
| Fig. 1. (a) XPS survey spectra, (b) high-resolution N 1s, (c) corresponding ratios of different doped N species, and (d) high-resolution Co 2p spectra of Co-N-C | |
{kind=link}
The Raman spectra of Co-N-C are presented in Fig. S3b (Supporting information). These spectra displayed two strong peaks at 1353 and 1580 cm-1, assigned to the D and G bands, respectively. The D band was ascribed to the disordered sp3- hybridized carbons [40] or defects such as edges and boundaries [58, 59], whereas the G band was associated with the ordered graphitized carbons or the sp2-hybridized carbon atoms [59-61]. The intensity ratio of the D band to G band (ID/IG) of Co-N-C-700, -800 and -900 were 1.95, 1.84 and 1.58, respectively. The value of ID/IG decreased with increasing pyrolysis temperature, indicating that the size of the sp2 domains within Co-N-C increased or the defects within Co-N-C decreased with the increase of pyrolysis temperature. This result was consistent with the XPS results that the doped N content within Co-N-C reduced and thus the conjugated domains enhanced when the pyrolysis temperature rose.
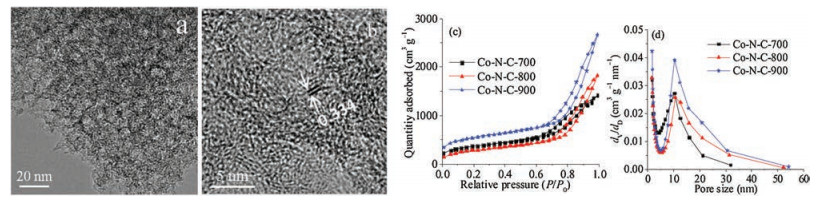
The morphology and microstructure of Co-N-C materials were investigated by SEM, TEM, and Accelerated Surface Area and Porosimetry System. The SEM images (Fig. S4 in Supporting information) demonstrated that three Co-N-C materials consisted of many nanoparticles. The TEM images of Co-N-C-800 are displayed in Figs. 2a and b. In Fig. 2a, Co-N-C-800 exhibited a great many mesopores with a diameter of ~8 nm, and the carbon shell thickness of the mesopores was ~3 nm. The diameter of the mesopores was accordant with that (~8 nm) of SiO2 nanoparticle template (Fig. S5 in Supporting information). In Fig. 2b, the interlayer spacing of carbon is clearly exhibited, which was ~0.334 nm. Additionally, the N and Co elements were homogeneously distributed in the carbon skeleton (Fig. S6 in Supporting information). Figs. 2c and d are the nitrogen sorption isotherms and pore size distribution curves, respectively. The three Co-N-C materials exhibited the typical IV isotherms with hysteresis loops in the high-pressure region, indicating the existence of welldefined mesopores. The pore-size distribution curves revealed that they possessed the hierarchical porous structure involving mesopores and micropores. Moreover, the mesopores were centred at ~8 nm because they mainly originated from the etching of SiO2 nanoparticles with a diameter of ~8 nm (Fig. S5). The detailed data on the pore structure parameters are shown in Table S3 (Supporting information). Notably, these three Co-N-C materials exhibited a specific surface area higher than 1200 m2/g and a total pore volume larger than 1.9 cm3/g, which could supply the fast mass transport channels and plentiful active sites for the catalysis of ORR and HER [62, 63].

|
Download:
|
| Fig. 2. (a) TEM and (b) HRTEM images of Co-N-C-800, (c) Nitrogen sorption isotherms, (d) BJH pore size distribution of Co-N-C | |
{kind=link}
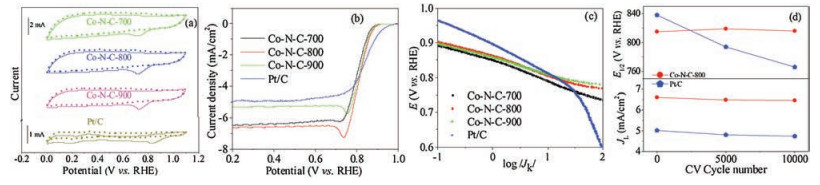
The cyclic voltammetry (CV), rotating disk electrode (RDE) and rotating ring-disk electrode (RRDE) measurements were applied to evaluate the ORR activity of Co-N-C and reference Pt/C catalyst in 0.1 mol/L KOH electrolyte. The CV tests were performed in both the O2- and N2-saturated KOH (Fig. 3a). All of the Co-N-C catalysts exhibited large quasi-rectangular double-layer capacities in the N2- saturated electrolyte, displaying excellent supercapacitor performance [50, 64]. For example, the capacity of C-N-Co-700 was 171 F/g, which was almost three times that of Pt/C (62 F/g) (Table S4 in Supporting information). Moreover, they denoted well-defined ORR peaks at 0.72 ~ 0.73 V in the O2-saturated electrolyte, showing excellent ORR activity. The RDE technique was then conducted to investigate the ORR activityofcatalysts. The LSV curves are exhibited in Fig. 3b, which were capacity-corrected by subtracting the background voltammograms results in the N2-purged electrolyte (Fig. S7a in Supporting information) [13, 65]. The half wave potential (E1/2) and limited current density (JL) are compiled in Table S4. The E 1/2 of Co-N-C-800 was 0.82 V at 1600 rpm, which was only 20 mV more negative than that (0.84 V) of Pt/C. However, the JL of Co-N-C- 800 was 6.62 mA/cm2, much higher than that (4.98 mA/cm2) of Pt/C. Moreover, Co-N-C-800 displayed the best ORR performance among the three Co-N-C catalysts. This mainly was because Co-N-C-800 showed the highest ratios of pyridinc and graphitic N to pyrrolic N and the highest Co contentamong the threeCo-N-C catalysts (Fig. 1c and Table S2). Fig. 3c shows the corresponding Tafel plots derived fromthecurvesinFig.3b, and their Tafel slope is exhibited in Fig.S7b in Supporting information.Atthe lowoverpotential region, theTafel slope of Co-N-C-800 was only 38 mV/dec, remarkably superior to thatof Pt/C catalyst (67 mV/dec), implying that Co-N-C-800 showed faster surface ORR rate than Pt/C catalyst [31]. In the high overpotential region, the Tafel slope of Co-N-C-800 was declined to 23 mV/dec, while that of Pt/C catalyst rapidly soared to 370 mV/dec. Such a opposite shifty tendency of their Tafel slopes in the high overpotential region suggested that Co-N-C-800 possessed much faster ORR rate than Pt/C catalyst because its hierarchicalpore structure and high specific surface area facilitated to transfer substanceand expose the abundant ORR activesites[31]. Asaresult, the ORR activity of Co-N-C-800 catalyst was comparable to Pt/C catalysts and higher than that of most previous transition metal/ nitrogen codoped carbons (Table S5 in Supporting information).

|
Download:
|
| Fig. 3. (a) CV curves of Co-N-C and Pt/C between 0 and 1.1 V in the O2- (solid line) and N2-saturated (dashed line) 0.1 mol/L KOH at 50 mV/s, (b) LSV curves of Co-N-C and Pt/C between 0 and 1.1 V in the O2-saturated 0.1 mol/L KOH at 10 mV/s and 1600 rpm, (c) Tafel curves derived from Fig. 3b, (d) Half-wave potential and limited current density of Co-N-C-800 and Pt/C as a function of CV cycle number in the O2-saturated 0.1 mol/L KOH solution | |
{kind=link}
The RDE curves of the Co-N-C and Pt/C catalysts at various rotation rateswere drawn and usedto analyze the mixture reaction kinetics on the basic of Koutecky-Levich (K-L) equations (Fig. S8 in Supporting information). The current density data at the potential ranging from 0.4 V to 0.7 V were collected, and the J-1 versus ω-1/2 plots were also drawn. All these plots represented excellent linear correlativity between J-1 and ω-1/2. As shown in Fig. S8, Co-N-C-700, -800 and -900 displayed a electron transfer number (n) of 3.77, 4.19 and 3.78 at 0.7 V, respectively, indicating that the catalysis process of ORR was nearly a four-electron pathway [41, 49, 50]. It was observed that Co-N-C-800 had the highest n, even higher than that (3.79) of Pt/C (Figs. S8d and h). The kinetic current density (Jk = 66.5 mA/cm2) of Co-N-C-800 at 0.7 V was larger than twice that (27.8 mA/cm2) of Pt/ C, ascribed to the hierarchical pore structure of Co-N-C-800 [31]. To further verify the reaction kinetics of Co-N-C and Pt/C, the RRDE measurements were carried out at 1600 rpm (Figs. S9a and c in Supporting information). Thus the corresponding hydrogen peroxide yields (H2O2%) and n could be directly calculated from the equation H2O2(%) = 200×(ir/N)/(ir/N+id) and n = 4×id/(ir/N+id) (Supporting information). As shown in Figs. S9b and d (Supporting information), the hydrogen species yield and n of Co-N-C-800 were 15.8% and 3.70 at 1.0 V, which were superior to the hydrogen peroxide yield (31.9%) and n (3.4) of Pt/C at 1.0 V, respectively.
Long-term stability and methanol tolerance were also the important properties of ORR catalysts. The Co-N-C-800 and Pt/C catalysts were cycled continuous 10, 000 cycles between 0.6–1.0 V at 50 mV/s in the O2-saturated 0.1 mol/L KOH electrolyte (Figs. S10a and b in Supporting information) [6]. As shown in Fig. 3d and Fig. S10a, the E1/2 and JL of Co-N-C-800 almost displayed no change, indicating excellent stability in 0.1 mol/L KOH electrolyte. By contrast, the Pt/C catalyst showed 72 mV loss of E1/2 and 0.28 mA/ cm2 loss of JL under the same conditions because of the loss of the Pt active sites during continuous operation (Fig. 3d and Fig. S10b)[6]. All the above results suggested that Co-N-C-800 showed an outstanding long-term stability, compared with Pt/C and the reported M-N-C catalysts [1, 11, 18]. Additionally, the methanol tolerance was investigated by CV measurements before and after adding 1 mol/L methanol in 0.1 mol/L KOH. The ORR peak of Co-NC-800 almost did not shift (Fig. S10c in Supporting information), implying that Co-N-C-800 possessed outstanding methanol tolerance. On the contrary, the ORR peak of Pt/C completely disappeared and a new methanol oxidation peak appeared (Fig. S10d in Supporting information). Hence, Co-N-C-800 exhibited much better methanol tolerance than Pt/C, especially suitable for the application in direct methanol fuel cells.
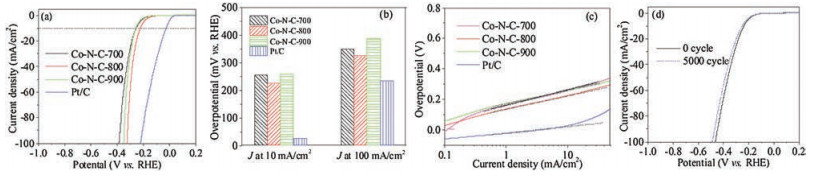
The HER catalytic activity of Co-N-C and Pt/C in 1.0 mol/L KOH are presented in Fig. 4. The overpotential of Co-N-C-700, -800 and -900 were 255, 224, and 258 mV at a current density of 10 mA/cm2 (Fig. 4a). It was obviously seen that Co-N-C-800 had the best HER activity among the three Co-N-C catalysts, but lower than that of Pt/C at an equal current density. However, the overpotential difference value between Co-N-C-800 and Pt/C catalysts sharply decreased with increasing current density (Fig. 4b). At a high current density of 220 mA/cm2, Co-N-C-800 and Pt/C with an equal catalyst loading of 0.2 mg/cm2, displayed an equal overpotential, indicating that the HER activity of Co-N-C-800 was comparable with that of Pt/C catalyst at the high overpotential region (Fig. S11 in Supporting information). Moreover, the HER activity of Co-N-C- 800 was better than that of most M-N-C bifunctional catalysts reported previously (Table S5). The Tafel plots of the Co-N-C and Pt/ C catalysts are displayed in Fig. 4c. The Tafel slope of Co-N-C-700, -800 and -900 was 101, 88 and 90 mV per decade, respectively. The Tafel slope of Co-N-C-800 was the lowest one among the three CoN-C catalysts and lower than that of most transition metal/N codoped carbon catalysts (Table S5). Electrochemical stability was another important parameter of the HER catalytic performance. The HER polarization curves of Co-N-C-800 before and after 5000 cycles in 1.0 mol/L KOH were indicated in Fig. 4d. Its overpontential only denoted a little change, revealing the excellent stability of the HER activity of Co-N-C-800 in KOH medium [66]. Furthermore, the chronopotentiometry test was carried out to study the long-term stability of Co-N-C-800. As shown in Fig. S12 (Supporting information), the potential of Co-N-C-800 remained stable over a long time of 24 h at a constant current density of 10 mA/cm2, revealing the superior stability in the strong alkaline medium [67, 68]. Moreover, Co-N-C-800 showed low charge-transfer resistance (~6 V) in the electrochemical impedance spectroscopy [66], which contributed to fast charge transport and superior catalytic performance (Fig. S13 in Supporting information). As a result, Co-N-C-800 showed excellent HER performance as transition metal/N codoped carbon catalysts.

|
Download:
|
| Fig. 4. (a) HER curves of Co-N-C and Pt/C in the N2-saturated 1.0 mol/L KOH at 5 mV/s, (b) corresponding overpotential of Co-N-C and Pt/C at various J values, (c) Tafel plots derived from Fig. 4a, (d) initial and post-potential HER curves of Co-N-C-800 in 1.0 mol/L KOH | |
{kind=link}
To identify the contribution of Co element for the ORR and HER catalytic activity, the SCN- poison test was performed according to the previous method [13, 69, 70]. After adding 10 mmol/L KSCN into 0.1 mol/L KOH, the E1/2 of Co-N-C-800 decreased from 0.82 V to 0.81 V and JL reduced by 1.30 mA/cm2, which implied that the SCN- ions blocked the Co active sites and deactivated them for ORR catalysis (Fig. S14a in Supporting information). Similarly, the HER activity of Co-N-C-800 also markedly reduced during the SCN- poison test. Its overpotential increased from 224 mV to 256 mV at 10 mA/cm2 (Fig. S14b in Supporting information). These results illustrated that the Co species exhibited great contribution to the electrocatalytic ORR and HER.
In summary, P4VP and cobalt chloride hexahydrate were mixed to form coordination polymer and then pyrolyzed using SiO2 nanoparticles as hard template, fabricating Co/N codoped carbon materials. To our knowledge, these Co-N-C materials are the first M-N-C bifunctional electrocatalyst derived from the "noncarbonizable" polymers for ORR and HER. Co-N-C-800 exhibited excellent catalytic performances for ORR in alkaline electylte (E1/2 = 0.82 V, JL = 6.62 mA/cm2), comparable to that of Pt/C catalyst. Moreover, its HER catalytic performances in alkaline electrolyte were superior to those of most carbon based catalysts. In particular, its bifunctional electrocatalytic performances for ORR and HER were better than those of most bifunctional doped carbon catalysts in KOH electrolyte. This research paves a novel avenue to fabricate high performance M-N-C catalysts for ORR and HER by pyrolysis of metal ion coordinated "noncarbonizable" polymers.
AcknowledgmentsThis research was supported by the National Natural Science Foundation of China (Nos. 21774073, 51690151, 21404071, and 21320102006) and the National Basic Research Program (No.2016YFA0201500).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.09.017
| [1] |
M. Kuang, Q. Wang, P. Han, G. Zheng, Adv. Energy Mater. 7 (2017) 1700193-1700200. DOI:10.1002/aenm.201700193 |
| [2] |
X.X. Liu, J.B. Zang, L. Chen, et al., J. Mater. Chem. A 5 (2017) 5865-5872. DOI:10.1039/C6TA10591B |
| [3] |
D.K. Singh, R.N. Jenjeti, S. Sampath, M. Eswaramoorthy, J. Mater. Chem. A 5 (2017) 6025-6031. DOI:10.1039/C6TA11057F |
| [4] |
L. Yang, J. Yu, Z. Wei, et al., Nano Energy 41 (2017) 772-779. DOI:10.1016/j.nanoen.2017.03.032 |
| [5] |
X. Jia, Y. Zhao, G. Chen, et al., Adv. Energy Mater. 6 (2016) 1502585-1502590. DOI:10.1002/aenm.201502585 |
| [6] |
H.W. Liang, X. Zhuang, S. Bruller, X. Feng, K. Müllen, Nat. Commun. 5 (2014) 4973-4979. DOI:10.1038/ncomms5973 |
| [7] |
X. Cai, B.Y. Xia, J. Franklin, et al., J. Mater. Chem. A 5 (2017) 2488-2495. DOI:10.1039/C6TA09615H |
| [8] |
Y. Hao, Y. Xu, N. Han, J. Liu, X. Sun, J. Mater. Chem. A 5 (2017) 17804-17810. DOI:10.1039/C7TA03996D |
| [9] |
S. Li, B. Li, L. Ma, J. Yang, H. Xu, Chin. Chem. Lett. 28 (2017) 2159-2163. DOI:10.1016/j.cclet.2017.08.029 |
| [10] |
J. Balamurugan, S.G. Peera, M. Guo, et al., J. Mater. Chem. A 5 (2017) 17896-17908. DOI:10.1039/C7TA04807F |
| [11] |
L. Liu, S. Ci, L. Bi, J. Jia, Z. Wen, J. Mater. Chem. A 5 (2017) 14763-14774. DOI:10.1039/C7TA04114D |
| [12] |
Z. Wang, H. Lei, R. Cao, M. Zhang, Electrochim. Acta 171 (2015) 81-88. DOI:10.1016/j.electacta.2015.05.004 |
| [13] |
Z. Pei, Z. Tang, Z. Liu, M. Zhu, D. Tang, C. Zhi, et al., J. Mater. Chem. A 6 (2018) 489-497. DOI:10.1039/C7TA09254G |
| [14] |
Z. Zhang, Z. Yi, J. Wang, et al., J. Mater. Chem. A 5 (2017) 17064-17072. DOI:10.1039/C7TA03999A |
| [15] |
Y. Zhao, C. Chang, F. Teng, et al., Adv. Energy Mater. 7 (2017) 1700005-1700011. DOI:10.1002/aenm.201700005 |
| [16] |
X. Fan, Z. Peng, R. Ye, H. Zhou, X. Guo, ACS Nano 9 (2015) 7407-7418. DOI:10.1021/acsnano.5b02420 |
| [17] |
R. Wang, X.Y. Dong, J. Du, J.Y. Zhao, S.Q. Zang, Adv. Mater. (2017) 1703711-1703720.
|
| [18] |
K. Shijina, R. Illathvalappil, S. Kurungot, et al., ChemistrySelect 2 (2017) 8762-8770. DOI:10.1002/slct.201701416 |
| [19] |
J. Yang, X. Wang, B. Li, et al., Adv. Funct. Mater. 27 (2017) 1606497-1606507. DOI:10.1002/adfm.v27.17 |
| [20] |
Y. Liu, F. Chen, W. Ye, et al., Adv. Funct. Mater. 27 (2017) 1606034-1606039. DOI:10.1002/adfm.v27.12 |
| [21] |
S. Cao, N. Han, J. Han, et al., ACS Appl. Mater. Interfaces 8 (2016) 6040-6050. DOI:10.1021/acsami.5b11955 |
| [22] |
J.J. Xu, C.H. Xiao, S.J. Ding, Chin. Chem. Lett. 28 (2017) 748-754. DOI:10.1016/j.cclet.2016.12.006 |
| [23] |
D. Ji, S. Peng, J. Lu, et al., J. Mater. Chem. A 5 (2017) 7507-7515. DOI:10.1039/C7TA00828G |
| [24] |
Y. Hao, Y. Xu, W. Liu, X. Sun, Mater. Horiz. 5 (2018) 108-115. DOI:10.1039/C7MH00706J |
| [25] |
T. Meng, J. Qin, S. Wang, et al., J. Mater. Chem. A 5 (2017) 7001-7014. DOI:10.1039/C7TA01453H |
| [26] |
I.S. Amiinu, Z. Pu, X. Liu, et al., Adv. Funct. Mater. 27 (2017) 1702300-1702310. DOI:10.1002/adfm.v27.44 |
| [27] |
X. Li, H. Lei, X. Guo, et al., ChemSusChem 10 (2017) 4632-4641. DOI:10.1002/cssc.201701196 |
| [28] |
H. Sun, Y. Han, H. Lei, M. Chen, R. Cao, Chem. Commun. 53 (2017) 6195-6198. DOI:10.1039/C7CC02400B |
| [29] |
G. Wan, C. Yang, W. Zhao, J. Shi, et al., Adv. Mater. 29 (2017) 1703436-1703443. DOI:10.1002/adma.201703436 |
| [30] |
H.C. Huang, I. Shown, S.T. Chang, et al., Adv. Funct. Mater. 22 (2012) 3500-3508. DOI:10.1002/adfm.v22.16 |
| [31] |
W. He, C. Jiang, J. Wang, L. Lu, Angew. Chem. Int. Ed. 53 (2014) 9503-9507. DOI:10.1002/anie.201404333 |
| [32] |
Y. Jiao, Y. Zheng, M. Jaroniec, S.Z. Qiao, Chem. Soc. Rev. 44 (2015) 2060-2086. DOI:10.1039/C4CS00470A |
| [33] |
N. Fu, H.M. Wei, H.L. Lin, et al., ACS Appl. Mater. Interfaces 9 (2017) 9955-9963. DOI:10.1021/acsami.6b15723 |
| [34] |
F.Q. Liu, W. Li, J. Zhao, et al., J. Mater. Chem. A 3 (2015) 12252-12258. DOI:10.1039/C5TA01536G |
| [35] |
K. Nasouri, A.M. Shoushtari, M.R.M. Mojtahedi, Polym. Compos. 38 (2017) 2026-2034. DOI:10.1002/pc.v38.9 |
| [36] |
J. Shi, X. Zhou, P. Xu, et al., Electrochim. Acta 145 (2014) 259-269. DOI:10.1016/j.electacta.2014.08.091 |
| [37] |
C. You, D. Dang, X. Qiao, et al., J. Mater. Chem. A 3 (2015) 23512-23519. DOI:10.1039/C5TA05599G |
| [38] |
M. Li, T. Liu, X. Bo, et al., J. Mater. Chem. A 5 (2017) 5413-5425. DOI:10.1039/C6TA09976A |
| [39] |
B. Wang, L. Xu, G. Liu, et al., J. Mater. Chem. A 5 (2017) 20170-20179. DOI:10.1039/C7TA05002J |
| [40] |
X. Lu, Z. Li, X. Yin, et al., Int. J. Hydrogen Energy 42 (2017) 17504-17513. DOI:10.1016/j.ijhydene.2017.02.090 |
| [41] |
Y. Zhang, J. Ge, L. Wang, et al., Sci. Rep. 3 (2013) 2771-2778. DOI:10.1038/srep02771 |
| [42] |
X. Ren, J. Liu, X. Meng, et al., Chem.-Asian J. 9 (2014) 1054-1059. DOI:10.1002/asia.v9.4 |
| [43] |
K. Parvez, S.B. Yang, Y. Hernandez, et al., ACS Nano 6 (2012) 9541-9550. DOI:10.1021/nn302674k |
| [44] |
H.W. Liang, W. Wei, Z.S. Wu, X. Feng, K. Mullen, J. Am. Chem. Soc. 135 (2013) 16002-16005. DOI:10.1021/ja407552k |
| [45] |
R. Silva, D. Voiry, M. Chhowalla, T. Asefa, J. Am. Chem. Soc. 135 (2013) 7823-7826. DOI:10.1021/ja402450a |
| [46] |
Y. Li, W. Zhou, H. Wang, et al., Nat. Nanotech. 7 (2012) 394-400. DOI:10.1038/nnano.2012.72 |
| [47] |
D. Villers, X. Jacques-Bédard, J.P. Dodelet, J. Electrochem. Soc. 151 (2004) A1507-A1515.
|
| [48] |
M. Sevilla, Valle-Vigón P., A.B. Fuertes, Adv. Funct. Mater. 21 (2011) 2781-2787. DOI:10.1002/adfm.201100291 |
| [49] |
J. Zhang, L. Qu, G. Shi, et al., Angew. Chem. Int. Ed. 55 (2016) 2230-2234. DOI:10.1002/anie.201510495 |
| [50] |
L. Lin, Q. Zhu, A.W. Xu, J. Am. Chem. Soc. 136 (2014) 11027-11033. DOI:10.1021/ja504696r |
| [51] |
D. Shin, B. Jeong, B.S. Mun, et al., J. Phys. Chem. C 117 (2013) 11619-11624. DOI:10.1021/jp401186a |
| [52] |
U.I. Koslowski, I. Herrmann, P. Bogdanoff, et al., ECS Trans. 13 (2008) 125-141. |
| [53] |
G. Wu, C.M. Johnston, N.H. Mack, et al., J. Mater. Chem. 21 (2011) 11392-11405. DOI:10.1039/c0jm03613g |
| [54] |
W. Xia, C. Qu, Z. Liang, et al., Nano Lett. 17 (2017) 2788-2795. DOI:10.1021/acs.nanolett.6b05004 |
| [55] |
J. Zhu, A.S. Childress, M. Karakaya, et al., Adv. Mater. 28 (2016) 7185-7192. DOI:10.1002/adma.201602028 |
| [56] |
A. Mulyadi, Z. Zhang, M. Dutzer, W. Liu, Y. Deng, Nano Energy 32 (2017) 336-346. DOI:10.1016/j.nanoen.2016.12.057 |
| [57] |
S.H. Ahn, X. Yu, A. Manthiram, Adv. Mater. 29 (2017) 1606534-1606543. DOI:10.1002/adma.201606534 |
| [58] |
L.K. Shrestha, R.G. Shrestha, Y. Yamauchi, et al., Angew. Chem. Int. Ed. 54 (2015) 951-955. DOI:10.1002/anie.201408856 |
| [59] |
J. Tang, J. Wang, L.K. Shrestha, K. Ariga, et al., ACS Appl. Mater. Interfaces 9 (2017) 18986-18993. DOI:10.1021/acsami.7b04967 |
| [60] |
J. Tang, N.L. Torad, R.R. Salunkhe, et al., Chem.-Asian J. 9 (2014) 3238-3244. DOI:10.1002/asia.201402629 |
| [61] |
J. Wang, Z.X. Wu, L.L. Han, et al., Chin. Chem. Lett. 27 (2016) 597-601. DOI:10.1016/j.cclet.2016.03.011 |
| [62] |
C. Zhu, H. Li, S. Fu, D. Du, Y. Lin, Chem. Soc. Rev. 45 (2016) 517-531. DOI:10.1039/C5CS00670H |
| [63] |
J. Zhang, H. Li, P. Guo, H. Ma, X.S. Zhao, J. Mater. Chem. A 4 (2016) 8497-8511. DOI:10.1039/C6TA01657J |
| [64] |
S. Yang, L. Zhi, K. Tang, et al., Adv. Func. Mater. 22 (2012) 3634-3640. DOI:10.1002/adfm.v22.17 |
| [65] |
S. Lee, M. Choun, Y. Ye, et al., Angew. Chem. Int. Ed. 54 (2015) 9230-9234. DOI:10.1002/anie.201501590 |
| [66] |
J. Park, H. Lee, Y.E. Bae, et al., ACS Appl. Mater. Interfaces 9 (2017) 28758-28765. DOI:10.1021/acsami.7b08786 |
| [67] |
J. Jiang, Q. Liu, C. Zeng, L. Ai, J. Mater. Chem. A 5 (2017) 16929-16935. DOI:10.1039/C7TA04893A |
| [68] |
Y. Jia, L. Zhang, A. Du, et al., Adv. Mater. 28 (2016) 9532-9538. DOI:10.1002/adma.201602912 |
| [69] |
Hai-Xia Z., W. Jun, Z. Qi, et al., Adv. Sustainable Syst. 1 (2017) 1700020-1700027. DOI:10.1002/adsu.v1.6 |
| [70] |
S. Wang, J. Qin, T. Meng, M. Cao, Nano Energy 39 (2017) 626-638. DOI:10.1016/j.nanoen.2017.07.043 |