 2019, Vol. 30
2019, Vol. 30 


b University of Science and Technology of China, Hefei 230029, China
α-Glucosidase (EC 3.2.1.20) plays an important role in the metabolic process of the body. α-Glucosidase, a membrane-bound enzyme located in the brush border membrane, can hydrolyze the complex carbohydrates into monosaccharides and the latter is absorbed into blood, thus causing postprandial hyperglycemia [1, 2]. α-Glucosidase inhibitors can delay the digestion of carbohydrates and absorption of monosaccharides, resulting in reduced postprandial glucose levels [3]. Therefore, they are widely used in the treatment of patients with type-2 diabetes mellitus (T2DM) [4]. Currently, drugs that treat diabetes in the market, such as acarbose [5], miglitol [6], usually cause gastrointestinal side effects. Thus, it is important to develop new α-glucosidase inhibitors with lower toxic and fewer adverse effects. The traditional Chinese medicines have been considered to be the most important source for enzyme inhibitors screening [7-9]. Many researches have shown that Polygonum cuspidatum possesses various beneficial effects including anti-infection, antidiabetes, anti-inflammation, protection against cardiovascular disease and cancer [10]. Polygonum cuspidatum is mainly composed of stilbene glycosides and anthraquinones [11]. Previous studies revealed that stilbene glycosides contribute to α-glucosidase inhibitory activity [12] and anthraquinones have been identified as the pharmacologically active constituents [13]. So it is great significant to screen α-glucosidase inhibitors from Polygonum cuspidatum.
Bioactivity-guided screening by free enzymes has been a main stream method in the discovery of active natural products. However, there are still some problems with this method, such as time-consuming and high-cost [14]. More importantly, it cannot be applied to complex sample screening due to its lack of separation ability [15]. However, immobilized enzyme technology can make up for these shortcomings [16, 17]. There are numerous advantages of immobilized enzymes including reusability, lowcost, easy handling, high efficiency separation and tolerance to changes in the environmental conditions [18, 19].
Immobilized carriers are critical for the development of immobilized enzyme. Nanoparticles have been receiving increasing attention in the field of immobilized enzymes due to their excellent properties, such as good biocompatibility, large specific surface area and easy modification [20, 21]. However, its property of small size is not conducive to the separation of immobilized enzymes from the reaction system [22]. Hopefully, magnetic nanoparticles (MNPs) can solve this problem very well, and they can be easily separated and controlled in an external magnetic field because of their inherent magnetic properties [23]. Therefore, MNPs have been developed and widely used as carriers for immobilized enzymes [24, 25]. It is difficult to directly immobilize the enzymes on the surface of MNPs, so several studies have focused on the surface modification to improve the stability against oxidation and dispersion of MNPs. For example, Asieh Soozanipour et al. [26] used silica-coated modified magnetite nanoparticles to immobilize xylanase, achieving high loading capacity, high catalytic activity and stability. Dongmei Liu et al. [22] immobilized α-glucosidase on the chitosan coated magnetic composites and screened enzyme inhibitors from traditional Chinese medicine and vegetables by capillary electrophoresis. However, there are still fewer reports about the immobilization of α-glucosidase on magnetic iron oxide nanoparticles.
In the past few decades, plenty of methods for enzyme inhibitors screening have been established, including colorimetric [27], fluorescenc eassay [28], capillary electrophoresis [7], electrochemical methods [29], HPLC [30], HPLC-MS/MS [31-33]. Among them, HPLC-MS/MS has attracted more and more attention for screening inhibitors from natural products owing to its advantages of sensitive and rapid detection, high efficiency separation, and low sample consumption.
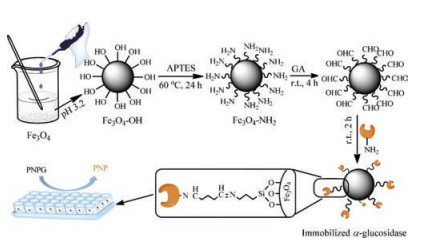
Therefore, the aim of current study is to prepare immobilized α- glucosidase using Fe3O4 magnetic nanoparticles (Fe3O4 MNPs) and apply it to rapid separate and screen potential α-glucosidase inhibitors from Polygonum cuspidatum extract by HPLC-MS/MS. Firstly, we use Fe3O4 MNPs as the carrier core with (3-aminopropyl) triethoxysilane (APTES) grafting by our approach, and then α-glucosidase was stereoscopically immobilized on the surface of Fe3O4 MNPs via Schiff base reaction using GA as a crosslinking agent. Subsequently, the properties of immobilized α-glucosidase, such as pH tolerance, thermo stability and reusability, are studied. Finally, the potential inhibitors from Polygonum cuspidatum are screened and identified in the present study. There are two primary advantages of this method: (1) Avoid the inactivation and poor operational stability caused by the enzymes leached out from supports, compared with the methods of adsorption and embedding; (2) Avoid the long separation process and ligands can be directly identified by combining with HPLC-MS/MS. The brief flowchart of the proposed approach is illustrated in Fig. 1.

|
Download:
|
| Fig. 1. Schematic illustration of preparation and activity determination of immobilized α-glucosidase. | |
Fe3O4 MNPs were synthesized by using the chemical coprecipitation method according to the literature [18, 34]. Because the hydroxyl of the Fe3O4 MNPs was able to react with the oxethyl side-chain of APTES, the surface of the Fe3O4 MNPs can be modified by the amino of APTES via stirring of Fe3O4 MNPs and APTES (1:3, mass ratios) in 50% (v/v) ethanol at 60 ℃ for 24 h [26]. The product was collected using an external magnet and washed several times with water and ethanol to remove any unbound APTES, and then dried in an oven at 40 ℃. The obtained sample was labeled as MNPs-NH2.
α-Glucosidase was covalently immobilized on MNPs-NH2 by Schiff base reaction between amino group and aldehyde group, using GA as a crosslinking agent [35]. The detailed experimental process can be found in Supporting information.
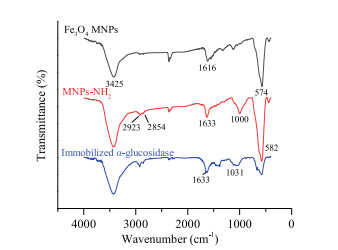
The surface modification and α-glucosidase binding were confirmed through the fourier transform-infrared spectroscopy (FT-IR) spectrograms and scanning electron microscopy (SEM). The results of FT-IR were shown in Fig. 2. The absorption peak at 3425 cm-1 in the spectrum of Fe3O4 MNPs was assigned to the O——H stretching vibration, whereas the peak at 1616 cm-1 corresponded to O——H deformed vibration. The characteristic adsorption peak of Fe——O stretching vibration was located at 574 cm-1. Compared with the FT-IR spectra of unmodified Fe3O4 MNPs, the IR spectra of MNPs-NH2 exhibited obvious adsorption peak of Si——O bending vibration at 1000 cm-1. Moreover, the adsorption peak of -CH2 and C——H stretching vibration appeared at 2923 cm-1 and 2854 cm-1. The adsorption band of stretching vibration of Fe——O at 574 cm-1 for Fe3O4 MNPs shifted to 582 cm-1 (MNPs-NH2) because of the interaction between Fe3O4 nanoparticles and APTES. These characteristics indicated that APTES had been successfully modified to the surface of Fe3O4 nanoparticles [36, 37]. The peak of Si——O bending vibration blue shift happened after immobilized α-glucosidase. In addition, all the adsorption peaks in the FT-IR spectra of immobilized α-glucosidase became less pronounced, especially the peaks of Si——O and Fe——O, compared with those in the FT-IR spectra of MNPs-NH2 [38]. Besides, the results of SEM (Fig. S1 in Supporting information) also indicated that the surface modification of every step was successful.

|
Download:
|
| Fig. 2. FT-IR spectra of Fe3O4 MNPs, MNPs-NH2 and immobilized α-glucosidase. | |
Immobilization conditions play important roles in obtaining the maximal enzyme activity. Therefore, we had optimized the four key parameters, including GA concentrations, ratio of MNPsNH2 to enzyme, crosslinking times and immobilization times.
Firstly, the effect of GA concentration on the relative enzyme activity was depicted in Fig. S2a (Supporting information). With the increase of GA concentration from 1% to 5%, the relative enzyme activity increased significantly. However, when GA concentration was more than 5%, the relative enzyme activity decreased. It might be that GA acted as a covalent cross-linker at low concentrations, while it was also used as a denaturing solvent at high concentrations [35]. So clearly, at low concentrations, the surface of nanoparticles had fewer active groups and therefore less enzyme was immobilized, resulting in lower relative enzyme activity. When the amount of enzyme immobilized on the surface of nanoparticles was saturated, the maximum relative enzyme activity was obtained at 5% of GA concentration. Excessive concentration of GA above its optimal value (5%) might result in the conformational changes of enzyme, decreasing the relative enzyme activity. Moreover, enzyme's flexibility was essential for its activity, so another reason for the decreased enzyme activity might be over cross-linking resulted in the rigidification of enzyme molecules [24].
Secondly, the influence of ratio of MNPs-NH2 to enzyme, which was controlled by fixing the amount of MNPs-NH2 and changing the enzyme concentrations, on the relative enzyme activity was investigated and shown as Fig. S2b (Supporting information). It was found that the relative enzyme activity firstly increased and then decreased with an increase of enzyme amount and when the ratio of MNPs-NH2 to enzyme was 32:1, maximum enzyme activity was obtained. The reason might be that the number of active groups in MNPs-NH2 modified GA (MNPs-GA) was certain, and when the free enzyme added was insufficient, the amount of enzyme immobilized on the surface of MNPs-GA was relatively less, which resulted in a lower enzyme activity. The relative activity increased with the increase of enzyme amount. When the amount of enzyme added was over saturated, α-glucosidase immobilized on the MNPs-GA was relatively crowded. The free enzyme would compete with immobilized α-glucosidase for the active site in the carrier, so that the active site was covered, causing a decrease in enzyme activity [37].
Thirdly, the relative enzyme activity reached its maximum when the immobilization time was 2 h, as shown in Fig. S2c (Supporting information). The result indicated that the amount of enzyme immobilized on the surface of nanoparticles was most suitable after immobilization for 2 h. When further increasing the immobilization time, the relative enzyme activity declined. This phenomenon was explained by the fact that α-glucosidase immobilized on the nanoparticles was too crowded to make the substrate easily access to the active site of enzyme, causing the decrease of relative acticity [22]. Besides, during the process of immobilization, α-glucosidase exposed to room temperature for a long time could also lose its activity.
In addition, the effect of crosslinking time on the relative activity can be found in Fig. S2d (Supporting information). When the crosslinking time was less than 4 h, enzyme activity increased with increasing time. And when the crosslinking time was more than 4 h, enzyme activity decreased with increasing time. This phenomenon was associated with amount of crosslinking agent linked to MNPs-NH2. The relative activity was lower within 4 h because the amount of GA cross-linked to MNPs-NH2 was less within a short time. Further increase of crosslinking time up to 8 h would result in decrease of relative activity, which might be on account of dimerization of GA. The prolonged contact time could reduce activity of GA, so that the amount of immobilized α- glucosidase was less, affecting the activity of enzyme [18].
Under the optimized conditions, the double-reciprocal plots of the free and immobilized α-glucosidase were constructed based on equation S1 and displayed in Fig. S3 (Supporting information). The Lineweaver-Burk plots were 1/v = (0.004370± 0.0001075) (1/ [S]) + (0.007955 ± 0.001351) and 1/v = (0.006183 ± 0.0001588) (1/ [S]) + (0.01559 ± 0.001996), respectively, for the free and immobilized α-glucosidase with determination coefficients of 0.9964 and 0.9961. The Michaelis-Menten constant Km is often used to evaluate the affinity of the enzyme to a specific substrate. The lower the value of Km, the higher is the affinity. In our experiment, Km values of the free and immobilized α-glucosidase were calculated to be 0.55 and 0.40 mmol/L, respectively. Km value of the free α-glucosidase was in agreement with the date found in literature [7], which was higher than that of immobilized α- glucosidase. It showed that the affinity of α-glucosidase to PNPG increased after immobilization. This might be explained by the fact that the better accessibility of the substrate to the active site of the immobilized enzyme and the milder diffusion barrier effects compared with free enzyme [39].
The pH tolerance, thermo stability and reusability of immobilized enzyme are important features in practical application. Fig. S4a (Supporting information) showed the pH tolerance result. The maximum enzyme activity of free α-glucosidase was achieved at 7.0, while pH optimum shifted from 7.0 to 7.5 after immobilization. The phenomena stated that the enzyme was more stable after immobilization. The reason was that α-glucosidase covalently binded to the surface of MNPs-GA via Schiff base reaction, which changed the conformation of the enzyme and increased its relative activity in a broad range of pH. In addition, the nanoparticles provided a buffering effect for the enzyme to improve the pH tolerance under alkaline or acidic conditions [37].
The thermo stability of the free and immobilized α-glucosidase were investigated and showed in Fig. S4b (Supporting information). Both of them displayed the same trend with the change of temperature. The higher the temperature was, the more obvious the advantage of immobilized α-glucosidase. Especially when the enzyme was placed at 47 ℃ for 1 h, the activity of free α- glucosidase was almost lost and only remained 1.23% of initial activity, whereas the immobilized α-glucosidase retained 19.23% of its initial activity. The result could be attributed to the immobilized α-glucosidase was more stable and more rigid by the multipoint binding between enzyme and the carrier compared with the free one [40].
The reusability was displayed in Fig. S4c (Supporting information). The immobilized α-glucosidase retained 66.0% of its initial activity after 7 cycles of reuse. The enzyme activity decreased with increasing frequency of use and the decrease in enzyme activity of the immobilized α-glucosidase could be attributed to inactivation during each cycle [38].
The application of immobilized α-glucosidase was examined by screening enzyme inhibitors from Polygonum cuspidatum extract. Firstly, we used acarbose, a positive inhibitor, to test the immobilized α-glucosidase and the result was presented in Fig. S5 (Supporting information). Then we screened inhibitors from Polygonum cuspidatum extract using HPLC-MS/MS. Fig. S6a (Supporting information) demonstrated the screen ability of immobilized α-glucosidase. In order to exclude false positives, we chose the blank Fe3O4 MNPs as a negative control. As can be seen from Fig. S6b (Supporting information), the signal intensity of eight components in blank Fe3O4 MNPs was much less than that of immobilized α-glucosidase. Therefore, the eight compounds screened were considered to be inhibitors of α-glucosidase. Next, the structures of eight compounds screened were identified by analyzing MS2 fragments data and comparing with MS data of standard compounds. The data related to eight compounds were listed in Table 1.
|
|
Table 1 HPLC retention times and MS characteristics of the active compounds of Polygonum cuspidatum extract incubated with immobilized α-glucosidase. |

{kind=link}
{kind=link}
In this study, we proposed a facile method to immobilize α- glucosidase on Fe3O4 MNPs modified with APTES. Immobilized α- glucosidase showed slightly improvement in pH tolerance and thermo stability compared with the free one. In addition, immobilized α-glucosidase displayed good reusability and the remanent activity still reached 66.0% after 7 recycles. It is an economical, convenient, and efficient technique for screening enzyme inhibitors when combined with HPLC-MS/MS. With the immobilized α-glucosidase, eight inhibitors were screened and identified from Polygonum cuspidatum extract. Hopefully, the immobilized α-glucosidase will be useful for diverse biotechnological applications, especially in pharmaceutical and medical application fields.
AcknowledgmentThis work was funded by the National Natural Science Foundation of China (Nos. 81473537, 81773690, 21673219).
Appendix A. Supplementary dataSupplementarymaterial related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.12.003.
| [1] |
H. Gao, Y.N. Huang, B. Gao, et al., Food Chem. 106 (2008) 1195-1201. DOI:10.1016/j.foodchem.2007.07.064 |
| [2] |
O'Keefe J.H., D.S.H. Bell, Am. J. Cardiol. 100 (2007) 899-904. DOI:10.1016/j.amjcard.2007.03.107 |
| [3] |
C. Schnack, G. Röggla, A. Luger, G. Schernthaner, Eur. J. Clin. Pharmacol. 30 (1986) 417-419. DOI:10.1007/BF00607953 |
| [4] |
M. Hanefeld, R.G. Josse, J.L. Chiasson, Diabetes Care 28 (2005) 1840-1841. DOI:10.2337/diacare.28.7.1840 |
| [5] |
J.A. Balfour, D. McTavish, Drugs 46 (1993) 1025-1054. DOI:10.2165/00003495-199346060-00007 |
| [6] |
J.P. Sels, M.S. Huijberts, B.H. Wolffenbuttel, Expert Opin. Pharmacother. 1 (1999) 149-156. DOI:10.1517/14656566.1.1.149 |
| [7] |
D.M. Liu, J. Chen, Y.P. Shi, Talanta 164 (2017) 548-555. DOI:10.1016/j.talanta.2016.12.028 |
| [8] |
D. Tang, J.X. Zhu, A.G. Wu, et al., J. Chromatogr. A 1286 (2013) 102-110. DOI:10.1016/j.chroma.2013.02.058 |
| [9] |
W.J. Chen, Y. Wu, X. Zhao, et al., Chin. Chem. Lett. 27 (2016) 1701-1707. DOI:10.1016/j.cclet.2016.05.022 |
| [10] |
Y. Zhao, M.X. Chen, K.T. Kongstad, A.K. Jager, D. Staerk, J. Agric. Food Chem. 65 (2017) 4421-4427. DOI:10.1021/acs.jafc.7b01353 |
| [11] |
Z. Pan, H. Liang, C. Liang, W. Xu, Chin. J. Chromatogr. 33 (2015) 22-28. DOI:10.3724/SP.J.1123.2014.07006 |
| [12] |
F. Li, Z. Zhan, F. Liu, et al., Org. Lett. 15 (2013) 674-677. DOI:10.1021/ol3035033 |
| [13] |
Z. Uddin, Y.H. Song, Curtis-Long M.J., et al., J. Ethnopharmacol. 193 (2016) 283-292. DOI:10.1016/j.jep.2016.08.026 |
| [14] |
E. Chaita, E. Gikas, N. Aligiannis, Phytochem. Anal. 28 (2017) 125-131. DOI:10.1002/pca.v28.2 |
| [15] |
Y.T. Zhu, X.Y. Ren, L. Yuan, et al., Food Chem. 173 (2015) 521-526. DOI:10.1016/j.foodchem.2014.10.087 |
| [16] |
E.M. Forsberg, J.R. Green, J.D. Brennan, Anal. Chem. 83 (2011) 5230-5236. DOI:10.1021/ac200534t |
| [17] |
J. Dong, W. Ning, W. Liu, M.L. Bruening, Analyst 142 (2017) 2578-2586. DOI:10.1039/C7AN00778G |
| [18] |
S. Talekar, A. Joshi, S. Kambale, et al., Chem. Eng. J. 325 (2017) 80-90. DOI:10.1016/j.cej.2017.05.054 |
| [19] |
Y. Rui, X.M. Wu, B.D. Ma, Y. Xu, Chin. Chem. Lett. 29 (2018) 1387-1390. DOI:10.1016/j.cclet.2017.10.033 |
| [20] |
M. Asgher, S. Noreen, M. Bilal, Int. J. Biol. Macromol. 95 (2017) 54-62. DOI:10.1016/j.ijbiomac.2016.11.012 |
| [21] |
Z. Fathi, E. Doustkhah, S. Rostamnia, et al., Int. J. Biol. Macromol. 117 (2018) 218-224. DOI:10.1016/j.ijbiomac.2018.05.156 |
| [22] |
D.M. Liu, J. Chen, Y.P. Shi, Int. J. Biol. Macromol. 105 (2017) 308-316. DOI:10.1016/j.ijbiomac.2017.07.045 |
| [23] |
W. Feng, J. Qiao, J. Jiang, et al., Talanta 182 (2018) 600-605. DOI:10.1016/j.talanta.2018.02.029 |
| [24] |
M.R. Ladole, J.S. Mevada, A.B. Pandit, Bioresource Technol. 239 (2017) 117-126. DOI:10.1016/j.biortech.2017.04.096 |
| [25] |
T. Wang, D. Li, B. Yu, J. Qi, J. Sep. Sci. 40 (2017) 1877-1886. DOI:10.1002/jssc.v40.9 |
| [26] |
A. Soozanipour, Taheri-Kafrani A., Landarani Isfahani A., Chem. Eng. J. 270 (2015) 235-243. DOI:10.1016/j.cej.2015.02.032 |
| [27] |
X.C. Li, X. Fan, L.J. Han, et al., Chin. J. Mar. Drugs 21 (2002) 8-11. |
| [28] |
P. Sledz, S. Lang, C.J. Stubbs, C. Abell, Angew. Chem. Int. Ed. 51 (2012) 7680-7683. DOI:10.1002/anie.201202660 |
| [29] |
M. Wang, Z. Xu, L. Chen, H. Yin, S. Ai, Anal. Chem. 84 (2012) 9072-9078. DOI:10.1021/ac301620m |
| [30] |
T. Matsui, T. Ueda, T. Oki, et al., J. Agric. Food Chem. 49 (2001) 1948-1951. DOI:10.1021/jf001251u |
| [31] |
Y. Tao, Y. Zhang, Y. Wang, Y. Cheng, Anal. Chim. Acta 785 (2013) 75-81. DOI:10.1016/j.aca.2013.04.058 |
| [32] |
Y. Tao, Z. Chen, Y. Zhang, Y. Wang, Y. Cheng, J. Pharm. Biomed. Anal. 78- 79 (2013) 190-201. |
| [33] |
J. Cao, J.J. Xu, X.G. Liu, S.L. Wang, L.Q. Peng, J. Chromatogr. A 1468 (2016) 86-94. DOI:10.1016/j.chroma.2016.09.022 |
| [34] |
X. Wang, L. Wang, X. He, Y. Zhang, L. Chen, Talanta 78 (2009) 327-332. DOI:10.1016/j.talanta.2008.11.024 |
| [35] |
S. Zhang, D. Wu, H. Li, et al., Food Funct. 8 (2017) 3219-3227. DOI:10.1039/C7FO00928C |
| [36] |
W. Hu, Z. Lou, Chem. Res. 24 (2013) 144-148. |
| [37] |
J.C. Feng, S.R. Yu, J. Li, T. Mo, P. Li, Chem. Eng. J. 286 (2016) 216-222. DOI:10.1016/j.cej.2015.10.083 |
| [38] |
D.M. Liu, J. Chen, Y.P. Shi, Anal. Chim. Acta 1006 (2018) 90-98. DOI:10.1016/j.aca.2017.12.022 |
| [39] |
D.M. Liu, J. Chen, Y.P. Shi, RSC Adv. 5 (2015) 56841-56847. DOI:10.1039/C5RA07982A |
| [40] |
T. Li, S. Li, N. Wang, L. Tain, Food Chem. 109 (2008) 703-708. DOI:10.1016/j.foodchem.2008.01.012 |