 2019, Vol. 30
2019, Vol. 30 


b State Key Laboratory of Explosion Science & Technology, Beijing Institute of Technology, Beijing 100081, China
It is well known that enantiopure chiral drugs could perform much different pesticide effects due to their corresponding specific structures. In order to obtain these optical pure enantiomers, chiral separation with high performance liquid chromatography (HPLC) has been extensively utilized either in scientific research or application areas. As the heart of one HPLC system, the choosing and optimization of chiral stationary phases (CSPs) has always been the top priority without doubt.
In the past years, one of the supramolecular compounds, cyclodextrin, especially β-cyclodextrin has attracted increasing attention using for chiral selector in HPLC & GC due to its unique structure of external hydrophilic and internal hydrophobic [1-3]. Abundant β-cyclodextrin derivatives have emerged in recent years to satisfy various separation demands. Among these derived groups, certain polar groups [4-8] provide new hydrogen bond sites, which contribute to the separation of substances whose chiral centers are distant from the plane of cyclodextrin inclusion complexes. In recent years, the research of ionic liquid has made a lot of achievements, its modification to the cyclodextrin enhances the electrostatic interaction within subject and object molecular thus perform good separation effects for polar compounds.
In general, as the supporter of the chiral selector, common silica gel is usually bonded to the β-cyclodextrin derivatives to obtain CSPs. This underived silica gel possesses strong water absorbability, which probably absorbs trace amounts of moisture in the air or solvent and form a polar water film on its surface. This hydration membrane will prolong the column equilibration time and diminish reproducibility of separation performance in various chromatographic modes. In 1989, J.J Pesek and coworkers [9] first reported modified silica gel whose surface is covered by silicahydrogen bond instead of silica-hydroxyl, the hydride silica. The substitution of hydroxyl eliminates this negative influence of water film, improving the separation rate and also the scope of solvents. Moreover, compared to ordinary silica, hydride silica possesses a higher stability and smaller polarity. It has been demonstrated that hydride silica can be stored under inert condition for 10 years without Si-H bond deterioration. In the following twenty years, much work has been done for the preparation and application of hydride silica [10-12]. It was initially prepared by replacing the hydroxyl groups with SOCl2 to chlorine and then reducing it to proton with an inorganic hydrogenation reagent LiAlH4 [13]. However, rigorous anhydrous condition and long reaction period has made it difficult for the synthesis. In 1993 [14], J.J Pesek first obtained hydride silica whose proton coverage was up to 95% by covering the hydroxyl groups with silanization reagent (EtO)3SiH (TES) with an reaction time of 4 h, thus becoming the most common method for hydride silica preparation due to its efficiency and safety. The surface modification of hydride silica is mainly realized by constructing a Si-C bond through the hydrosilylation reaction, terminal olefin is the most commonly used hydrosilylation reagent.
In recent years, with the continuous development of organofluorine chemistry, fluorinated compounds have been playing an increasingly important role in the fields of medicine, agriculture and materials science [15, 16]. It is generally acknowledged that fluorine-containing functional groups in biologically active molecules can largely alter the physiochemical properties of bioactive molecules through its steric hindrance effect and the lipophilic metabolic effects. Trifluoromethyl is the most lipophilic group ever known, which has proved to be pivotal in drug molecular design. With a large ratio of trifluoromethyl compound among organic fluorides, preparation and separation of this kind of compound with specific configuration is of great significance to construct new medicine molecules and improve the bioactivity of drug molecules. Various pyrrolidine compounds have been prepared [17-20] in our previous work, here more than thirty racemates were injected as analytes for separation.
These chiral stationary phases were synthesized under laboratory conditions and all of the chemicals used were analytical grade. β-Cyclodextrin was recrystallized twice with water and dried at 100 ℃ for 12 h before use. p-Toluenesulfonyl chloride, triethoxysilane (TES), γ-(2, 3-epoxypropoxy)propytrimethoxysilane (KH-560), sodium hydride (60%) and azodiisobutyronitrile (AIBN) were purchased from commercial sources. Silica gel (5 mm, 100 Å) was purchased from FUJIK (Japan). DMF, pyridine, toluene, dioxane and tetrahydrofuran were dried with molecular sieve before use. Methanol and acetonitrile used as the mobile phase were HPLC grade. Analytes (ferrocene derivatives and chiral drugs) were prepared as previously reported.
With regard to the characterizations, melting points were determined on an XT4-100 apparatus. 1H NMR spectra were recorded in DMSO-d6 on a Bruker DRX-500/600 spectrometer. IR spectra were measured on a Bruker Nicolet Magna IR 560. MS were measured on a Bruker BRFLEX. Element analysis was performed on an Elementar Vario EL. The HPLC system used consisted of two Wellchrom HPLC pumps (K-501), a manual injection valve (model 7725i), a Wellchrom spectrophotometer (K-2501) and a dynamic mixing chamber.
The synthesis of the CSPs includes two sections, derivation of β- cyclodextrin and modification of silica. The detailed synthesis procedures of MHDNTCD, HPITCD, HPRTCD and HDNTCD CSPs are shown in Scheme 1. 1H NMR and mass spectrometry confirmed the generations of 5a, 5b, 5c. IR and elemental analysis were utilized to analyze the characteristics of these stationary phases. The concentrations of the derived β-cyclodextrins bonded to hydride silica were still calculated according to the formula [21]:
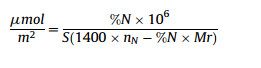
|

|
Download:
|
| Scheme 1. Synthesis routes for MHDNTCD, HPITCD, HPRTCD and HDNTCD CSPs. | |
where N is the percentage of nitrogen in the sample as determined by elemental analysis, Mr is the molecular weight of the CD derivative, nN is the number of nitrogen atoms in the chiral selector, and S is the special surface area of silica gel, which is 320 m2/g according to the manufacturer.
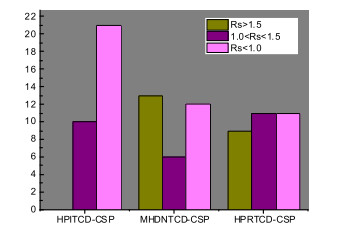
In order to estimate the comprehensive separation effect of these three CSPs, the separations of 35 racemic pyrrolidine compounds are summarized in Fig. 1. It is clear that the HPITCD-CSP which was modified by piperidine showed poorer separation effects compared to the other two CSPs. Almost no analytes obtained baseline separation (Rs>1.5) on HPITCD-CSP, most analytes (21) could not be separated completely (Rs < 1.0). While on both MHDNTCD-CSP and HPRTCD-CSP, most analytes were well separated. Especially on HPRTCD-CSP, in the same mobile phase mode, the separation effects under different composition ratios were all excellent, which was not achieved by the other two stationary phases under the same conditions.

|
Download:
|
| Fig. 1. The separation summary of three CSPs. | |
The average substitution degree of MHDNTCD was calculated as about 8. Compared to mono-substituted β-cyclodextrin, the interaction sites on chiral selector aiming to the analytes were greatly increased, the binding capacity was also stronger to improve its enantiomeric separation ability to some extent. Based on Table 1, most Rs values were higher than 1.8 for analytes who could be baseline separated on MHDNTCD-CSP, compound 7 obtained a highest Rs value of 6.43 under mobile phase composition of acetic acid/trimethylamine = 0.5/1. Moreover, compared to HPITCD-CSP and HPRTCD-CSP, higher capacity factors k were obtained in MHDNTCD-CSP, which ascribed to the high substitution, too. The combining force between analyte and chiral selector was also enhanced to have a high capacity of the components, making them strongly reserved and eluted slowly. The pH of mobile phase could bring influences to the separation as well as the stabilities of the CSPs, here acetic acid/triethylamine buffer solution was used to adjust the mobile phase pH under this chromatography mode. According to the data in Table 1, the composition of mobile phase affects obviously to the separation of analytes.
|
|
Table 1 Separation data for pyrrolidine compounds in polar-organic phase mode. |

Overall, the ratio change of the two components has little effect on the resolution of compounds 1, 2, 5-7, 9, due to their smaller polarity thus less affected by changes in the pH of the mobile phase. For compounds 12-15, 18-20, who possess strong polarity group, trifluoromethyl, in the 3-pyrrolidine, with the increase of acetic acid ratio in the mobile phase, the resolutions of majority of compounds show a decreasing tendency (as shown in Fig. 2). The addition of appropriate amount of acid or base in the mobile can bring a positive effect on the separation, while an excess of addition can increase the interaction between the analyte and the mobile phase. The strong polarity of fluorine makes it easy to form a firm hydrogen bond with protons in the mobile phase, which indirectly increases the contact between the analyte and the chiral selector, ensuring the chiral recognition process can be more fully realized. While on the other hand, the strong binding force makes the analyte difficult to be eluted, resulting in different degrees of negative impact on the separation results.

|
Download:
|
| Fig. 2. Rs values of compounds 18-21, 24, 28, 29, 31 under three pH of mobile phase conditions on column HPRTCD-CSP. | |
Except for pyrrolidine compounds, several chiral drugs have also been separated under this mode (Table 1). Different from the phenomenon that selectivity (α) and retention factors (k) varied slightly for most pyrrolidines while the ratio of acetic acid/ triethylamine changed, drug moleculars exhibited certain separation changes according to the different mobile phase compositions. For compound 32, when the ration of acetic acid/ trimethylamine = 1:1, the selectivity has reached to 2.93 on HPITCD-CSP and 1.97 on MHDNTCD-CSP, respectively. While even no separation could be realized on HPITCD-CSP but a baseline separation on MHDNTCD-CSP was observed. Though selectivity values obtained when acetic acid/trimethylamine = 1:0.5 or 0.5:1 were lower on HPITCD-CSP, 32 still could be separated successfully (with Rs values 1.33 and 2.47).
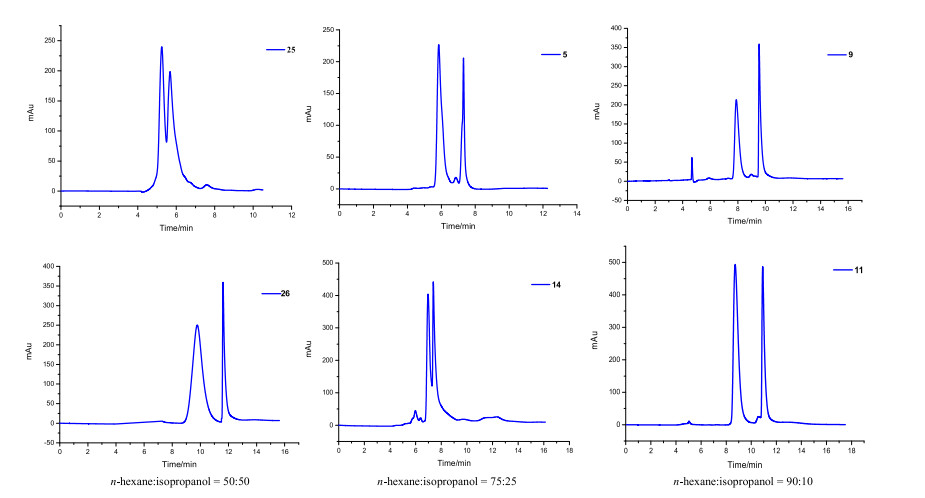
In our previous studies, these pyrrolidine compounds have been separated successfully under hexane-isopropanol mobile phase system using commercial chiral daicel OD-H, AD-H and AS-H columns, which inspired us to apply the new synthesized CSPs for the same separations. The composition of mobile phase were n-hexane/isopropanol = 90/10, 75/25, 50/50 (Table 2, Fig. 3).
|
|
Table 2 Separation data for pyrrolidine compounds and chiral drugs on MHDNTCD-CSP in normal phase mode. |

|
Download:
|
| Fig. 3. Chromatograms of compounds 5, 9, 11, 14, 25, 26 on MHDNTCD-CSP with three different compositions of mobile phase. | |
Generally speaking, the corresponding peak areas of two enantiomers in the separation chromatogram should be symmetrical in theory, since all the analytes separated were racemates. However, it was found that part of the analytes exihibited a peak area ratio of 60/40, even 70/30. With a chromatographic condition of HPRTCD-CSP as the solid phase, acetonitrile/methanol/acetic acid/triethylamine = 480/20/1/0.5 as the mobile phase, separations for single configuration exo-(2R, 3R, 4S, 5R) of compounds 18, 19, 21, 28-31 were selected for discussion.
According to Table 3, the retention times of seven single configuration compounds were all consistent with that of the first peak of their corresponding racemates, which indicated that exo- (2R, 3R, 4S, 5R) was eluted earlier than exo-(2R, 3S, 4R, 5R). Moreover, in terms of peak area, exo-(2R, 3R, 4S, 5R) also occupied a dominant ratio compared to its enantiomer. The reason is speculated as follows: The racemate was carried by mobile phase to the CSP and had interactions with the chiral selector through internal inclusion, external recognition as well as solvent molecules, finally achieved the distribution in two phases. During this process, the configuration of exo-(2R, 3S, 4R, 5R) was more easily to enter the cyclodextrin cavity and may had a stronger interaction with derived groups linked on the cyclodextrin, which provided a tighter combination between analyte and the chiral selector. In contrary, exo- (2R, 3R, 4S, 5R) was combined slightly to the selector, thus eluted earlier with a smaller retention time and a larger ratio of peak area.
|
|
Table 3 The retention time comparison between single configurations and their racemics. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Overall, three novel modified β-cyclodextrin derivatives and hydride silica were prepared and chemically bonded to obtain three modified chiral stationary phases. MHDNTCD-CSP exhibited excellent enantioselectivity for most analytes due to its high substitution and linked ionic groups. Moreover, the separations were extremely good on this stationary phase under normal phase, especially eluted with a low polarity mobile phase. The chiral center in HPRTCD-CSP ensures a proper steric configuration as well as stronger interaction between analytes and the chiral selector, which reduce the impact of mobile phase composition and provide a wide application range of pH. Since there is no p electrons contain in the derived group piperidine, only minority analytes were separated on HPITCD-CSP under specific mobile phase composition. An experimental and theoretical investigation were performed and discussed based on the shape problem of chromatograms, the steric configuration of enantiomers resulted in a stronger or weaker combination force as well as different configuration match between analytes and the CSPs, thus lead to the difference of elution degrees.
| [1] |
C.M. Lian, L.P. Jiang, D.L. Liu, Chin. Chem. Lett. 25 (2014) 134-136. DOI:10.1016/j.cclet.2013.10.025 |
| [2] |
M. Huang, W.J. Chen, Y. Zhou, et al., Chin. Chem. Lett. 24 (2013) 840-844. DOI:10.1016/j.cclet.2013.05.019 |
| [3] |
Q.J. Peng, J. Jiao, et al., Chin. Chem Lett. 25 (2014) 1416-1418. DOI:10.1016/j.cclet.2014.05.052 |
| [4] |
J.G. Yu, K.L. Huang, D.S. Huang, et al., Chin. Chem. Lett. 17 (2006) 1547-1550. |
| [5] |
X.P. Chen, Z.M. Zhou, H. Yuan, et al., Chin. Chem. Lett. 19 (2008) 797-800. DOI:10.1016/j.cclet.2008.04.045 |
| [6] |
G. Crini, B. Martel, G. Torri, et al., Chromatographia 41 (1995) 424-430. DOI:10.1007/BF02318617 |
| [7] |
A. Zhang, W. Lai, J. Sun, et al., J. Chromatogr. A 1281 (2013) 26-31. DOI:10.1016/j.chroma.2013.01.016 |
| [8] |
P. Zhang, G. Sun, K. Tang, et al., J. Sep. Sci. 37 (2014) 3443-3450. DOI:10.1002/jssc.v37.23 |
| [9] |
J.E. Sandoval, J.J. Pesek, Anal. Chem. 61 (1989) 2067-2075. DOI:10.1021/ac00193a013 |
| [10] |
J.J. Pesek, M.T. Matyska, P.F. Fu, Chromatographia 53 (2001) 635-640. DOI:10.1007/BF02493011 |
| [11] |
J.J. Pesek, M.T. Matyska, J.P. Venkat, J. Sep. Sci. 31 (2008) 2560-2566. DOI:10.1002/jssc.v31:14 |
| [12] |
J.J. Pesek, M.T. Matyska, K.V. Prajapati, J. Sep. Sci. 33 (2010) 2908-2916. DOI:10.1002/jssc.v33:19 |
| [13] |
J.E. Sandoval, J.J. Pesek, Anal. Chem. 63 (1991) 2634-2641. DOI:10.1021/ac00022a017 |
| [14] |
C.H. Chu, E. Jonsson, M. Auvinen, et al., Anal. Chem. 65 (1993) 808-816. DOI:10.1021/ac00054a027 |
| [15] |
J. Wang, M. Sánchezroselló, J.L. Aceña, et al., Chem. Rev. 114 (2014) 2432-2506. DOI:10.1021/cr4002879 |
| [16] |
L. He, W. Zhang, L. Zhao, et al., J. Chromatogr. A 1007 (2003) 39-45. DOI:10.1016/S0021-9673(03)00987-7 |
| [17] |
L. Dai, D. Xu, L.W. Tang, et al., Chemcatchem 7 (2015) 1078-1082. DOI:10.1002/cctc.201403048 |
| [18] |
L. Dai, X. Li, H. Yuan, et al., Tetrahedron. Asym. 22 (2011) 1379-1389. DOI:10.1016/j.tetasy.2011.07.025 |
| [19] |
L.W. Tang, B.J. Zhao, L. Dai, et al., Chem.-Asian J. 11 (2016) 2470-2477. DOI:10.1002/asia.201600941 |
| [20] |
L.W. Tang, B.J. Zhao, C. Li, et al., Tetrahedron 73 (2017) 923-930. DOI:10.1016/j.tet.2017.01.001 |
| [21] |
B.A. Siles, H.B. Halsall, J.G. Dorsey, J. Chromatogr. A 704 (1995) 289-305. DOI:10.1016/0021-9673(95)00269-S |