 2019, Vol. 30
2019, Vol. 30 


b School of Chemistry and Chemical Engineering, The Queen's University of Belfast, Belfast BT9 5AG, UK
Nitrogen oxides (NOx) generated by internal combustion engines cause many environmental issues. Currently, by virtue of the NOx remediation techniques, such as the three-way catalytic conversion (TWC), NOx storage/reduction (NSR) and selective catalytic reduction (SCR) [1-5], they can be largely removed under normal operating conditions (typically above 200 ℃). However, the corresponding low efficiency during cold starts remains a big problem. Particularly, in the large semi-closed space such as the road tunnels and indoor parking areas where cold starts occur frequently, tens of ppms of NOx (NO usually accounting for 90%) could accumulate as a result of incomplete NOx removal. Hence, the removal of low-content NO at room temperature serves as one of the important but challenging targets. Considering that NO2 can be readily absorbed in water or base solutions, to locate the highly efficient catalyst and achieve the catalytic oxidization of NO into NO2 at mild condition is a crucial solution.
Currently platinum-based catalysts [6-9] have been widely used for NO oxidation on relatively high temperature, and the high costs restrict their applications. Transition metal oxides (TMOs) have also attracted great attention as promising catalysts for NO oxidation, such as the La1-xSrxCoO3, La1-xSrxMnO3, and Mn-mullite (Sm, Gd)Mn2O5 [10-13]; however, they are still not satisfactory at the room temperature. The activated carbon, especially modified carbon [14] and activated carbon fibers [15], were reported to be active for NO oxidation at room temperature, but the activity diminished quickly because of the irreversible NO2 adsorption. Thus, exploring non-precious and room-temperature catalysts for dilute NO oxidation and understanding the catalytic mechanism are of great importance.
Recently, Cr-based catalysts [16-24] have drawn increasing attention owing to their superior ability in the complete oxidation of NO at relatively mild condition. Cai et al. studied the effect of Cr on the NO oxidation over the ceria-zirconia solid solutions, proposing that the well dispersed CrOx leads to much improved catalytic activity [20]. Liu et al. found that chromium oxides showed high NO to NO2 conversions at ambient temperature, and suggested that the amorphous phase structure of the prepared CrOx is critical in obtaining high performance and long-time stability for NO oxidation [22]. These attractive findings represent much progress, because Cr-based oxides are significantly less expensive and more abundant. However, several important issues on Cr-based catalysts remain to be addressed. Firstly, as a metal oxide with multivalence, it is still controversial for the real active component, resulting from various stoichiometric Cr-oxide phases could be formed during the fabrication process, including CrO3, Cr2O3 and even CrO2 phases. Wang et al. reported that CrOx-ZrO2 mixed oxide showed enhanced activity for the low-content NO oxidation at ambient-temperature, and suggested that the surface Cr6+ species is more favorable than Cr2O3 to oxidize NO [25]. Meanwhile, very recently Cai et al. observed the existence of the active CrO2 phase [26]. Theoretically, our previous work [27] have calculated the catalytic activities of Cr2O3 and the mono-chain CrO3 phase, finding that the Cr2O3(001) with the highly unsaturated Cr3c exposed exhibits strong adsorption ability and the overall activity is limited by the strong adsorption of NO2 species (poisoning effect), while on Cr2O3(012) exposing the fivecoordinated Cr5c, the reactant O2 cannot be efficiently adsorbed/ activated and also limits the catalytic ability for NO oxidation; moreover, the monochain-CrO3, which could be thermodynamically self-produced on Cr2O3(012), can give an enhanced NO oxidation activity in comparison with the bare Cr2O3(012) surface. Nevertheless, to the best of our knowledge, the catalytic ability of CrO2 phase for NO oxidation has been not explicitly known either experimentally or theoretically.
Secondly, the favorable reaction mechanism behind the roomtemperature NO oxidation on the CrO2 is not yet clear. The usually exposed (110) surface of CrO2 give two kinds of active sites, the coordinately unsaturated Crcus and the lattice oxygens, which can result in the reaction routes following the Langmuir-Hinshelwood mechanism occurring on Crcus sites alone or the Mars-van Krevelen (MvK) one with the lattice oxygen involved. Which one is preferred for the low-content NO oxidation at room temperature has been not understood. Moreover, the NO oxidation process possibly involves kinds of intermediate species, which thus constitute a complex reaction network. Overall, it is therefore highly desirable to systematically explore the possible elementary steps.
Inspired by these issues, in this work, the most exposed (110) surface of rutile-type CrO2 was adopted to investigate the reaction mechanism of NO oxidation by combing the first-principles density functional theory (DFT) calculations and microkinetic analysis. The optimal reaction route and the rate-determining step for the low-content NO oxidation at room temperature were quantitatively figured out. We believed these findings would provide a molecular-level insight into the activity origin and advantages/shortcomings of CrO2 oxide in catalyzing NO oxidation. All the calculations in this work were performed within the framework of density functional theory (DFT) using the Vienna ab initio simulation package (VASP) [28, 29]. Generalized gradient approximation (GGA) with Perdew-Burke-Ernzerhof (PBE) functional [30] was employed to describe the exchange and correlation energy (see details in the Supporting information).
As illustrated in Fig. 1a, the CrO2(110) surface (serving as the most stable surface of rutile-type CrO2) is mainly terminated by alternating rows of two-coordinated bridge oxygen (Obri) and fivecoordinated Cr5c linked by the three-coordinated in-plane O3c, in which the coordinatively unsaturated Cr5c cations (the Lewis acid site) and Obri anions are usually considered to be vital for the catalytic activity, providing the basic adsorption sites. Specifically, considering that the adjacent Cr5c-Cr5c distance on the (110) surface is 2.955 Å, which implies that the NO oxidation could proceed along the Cr5c sites, we first calculated the adsorption of key species X (X = NO, NO2 and O2) on the Cr5c site and examined the possibility following the Langmuir-Hinshelwood (L-H) mechanism to catalyze NO oxidation.

|
Download:
|
| Fig. 1. The (110) surface (top view) of rutile-type CrO2 catalyst (a) and the possible molecular adsorption structures on CrO2(110) for NO (b, c), NO2 (d-g), and O2 (h, i). Inserted value on the top right of each configuration is the corresponding adsorption energy (in eV). | |
It was found that NO can relatively strongly adsorb on the Cr5c site in a preferential tilted η1-N configuration (Fig. 1b, Eads = - 1.14 eV) in comparison with the upright one (Fig. 1c, Eads = - 0.88 eV). For NO2, on examining four possible η1-N-nitro, η1-O-nitro, μ2-N, O-nitrito and μ2-O, O'-nitrito adsorption configurations (Figs. 1d–g, respectively), the bidentate μ2-O, O'-nitrito configuration is identified as the most stable one, giving an Eads of -1.18 eV (versus -0.58/-0.76/-0.82 eV for the other threes (Figs. 1d-f, respectively). By contrast, O2 can only moderately adsorb at the Cr5c site in either σ-bond (Fig. 1h) or π-bond configuration (Fig. 1i), with the adsorption energy being -0.57 eV and -0.49 eV, respectively. Moreover, the dissociation of the adsorbed O2* into atomic O* at the Cr5c sites is endothermic by 0.72 eV for the more stable O2* in the σ-bond configuration, and the corresponding barrier is as high as 2.02 eV, implying the infeasibility of obtaining the atomic O* via O2 dissociation at room temperature. In other words, the reactant O2 has to participate in the subsequent reactions in the molecular state. Additionally, in light of the fact that the adsorption of NO/NO2 at the Cr5c site is much superior to that of O2 (-1.14 eV vs. -0.57/-0.49 eV), in reality O2 could be expected to exist in minority at the Cr5c sites, owing to the competitive adsorption effect between NO* and O2*. Therefore, one may expect the overall activity of NO oxidation following the L-H mechanism is unsatisfactory.
To give a more quantitative examination on the catalytic activity, the specific reaction network of NO oxidation with the L-H mechanism was explored on CrO2(110), as depicted in Scheme 1. At the adjacent Cr5c site around NO*, O2 can weakly co-adsorb with an adsorption energy of -0.24 eV (Fig. S1a in Supporting information), which is lower than the O2-only adsorption (-0.57 eV), hinting that the preadsorption of NO inhibits the O2 adsorption. They can then easily couple with each other into a more stable ON*OO* species (Fig. S1c in Supporting information) with a low barrier of 0.11 eV (Fig. S1b for the corresponding transition state (TS) with the forming O-N bond being 2.463 Å). Subsequently, the formed ON*OO* (IS') can break its O-O bond at a barrier of 0.81 eV (Fig. S1d for the TS), yielding the intermediate μ2-N, O-nitrito NO2* and atomic O*, which corresponds to a reaction enthalpy of -0.03 eV. The NO2* can readily desorb at an energy cost of 0.51 eV and form the first NO2 molecule. With respect to the remaining O* atom, it can readily react with another NO* adsorbed nearby to form the second μ2-N, O-nitrito NO2*, and this progress is almost spontaneous; the barrier is rather low (0.11 eV, see TS in Fig. S1h with the forming O-N bond of 2.000 Å), and the reaction is much exothermic by -1.22 eV. Finally, the second NO2* desorbs into the gas phase, completing the catalytic cycle.

|
Download:
|
| Scheme 1. Elementary steps of NO oxidation based on the Langmuir-Hinshelwood path on CrO2(110) surface. | |
As shown by Figs. S2 and S3 (Supporting information), the energy profile, as well as the estimated free energy profile with the entropy effects of the reactant NO/O2/NO2 molecule included (TΔ SNO, TΔSO2 and TΔSNO2 are 0.66 eV, 0.64 eV and 0.75 eV at T = 300 K, respectively; see details in Supporting information), were calculated, which reveal that the successive conversion from the coadsorbed NO* and O2* into two NO2 molecules can proceed smoothly without high barriers. However, the O2 adsorption is weak (the adsorption free energy (ΔGads) including the entropy effect at room temperature is + 0.40 eV), and would be hindered specially due to the competitive effect resulting from the stronger NO adsorption (ΔGads= -0.48 eV) on CrO2(110). Accordingly, the low coverage of O2* can be expected, which is quantitatively estimated to be only 4.85 ×10-12 from the microkinetic analysis later. Overall, the conversion rate of NO oxidation at the Cr5c sites only following the L-H mechanism is limited (see further the microkinetic results).
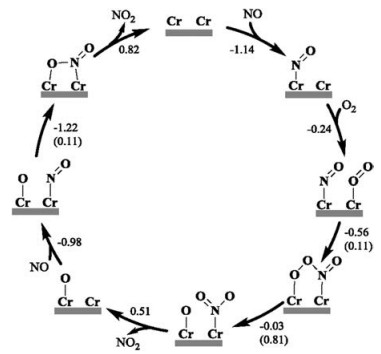
Owing to that the Cr5c site cannot activate O2 and provide reactive oxygen species to oxidize NO, we resorted to the participation of the surface lattice Obri in oxidizing NO, i.e., the Mars-van Krevelen (MvK) mechanism. As illustrated in Scheme 2a, it was found that NO* can easily couple with Obri without an apparent barrier (Ea = 0.01 eV) to form an ONObri (namely NO2#, # denoting the Ovac site) configuration (Fig. S4a in Supporting information) and the whole process is exothermic by -1.03 eV; in the optimized TS (Fig. S4b in Supporting information), the ON-Obri bond distance is 2.621 Å. This indicates that the Obri could be an important oxidative species in promoting NO oxidation on CrO2(110).

|
Download:
|
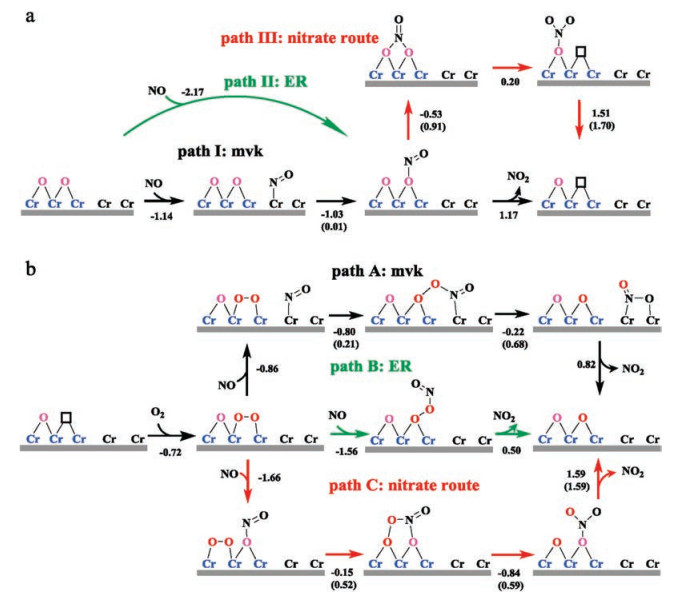
| Scheme 2. (a) Elementary steps of NO oxidation for the surface lattice Obri vacancy formed, based on three paths (black for path Ⅰ: the adsorbed NO* couples with Obri (Marsvan Krevelen path); green for path Ⅱ: NO directly couples with Obri from the gas phase (Eley-Rideal path); red for path Ⅲ: ONObri (NO2#) couples with the adjacent Obri to form the NO3 species (nitrate route) on CrO2(110). * and # denote the Cr5c and Ovac site, respectively. (b) Possible elementary steps of O2 adsorption and conversion to refill the Ovac. | |
Besides, it is worth discussing that the gaseous NO could also follow the Eley-Rideal mechanism to directly couple with Obri and form the ONObri species, being exothermic by 2.17 eV (path Ⅱ in Scheme 2a); even when the NO molecule is arbitrarily put around Obri at a long distance of ~3.0 Å, it was found that NO gradually descends and binds with Obri spontaneously upon a typical structural optimization, and thus this step could be a barrierless process in term of the total energy (without considering the entropy effect).
Afterwards, the formed ONObri (NO2#) could desorb with an Ovac left, i.e., NO2# → NO2 + Ovac, corresponding to a reaction enthalpy of 1.17 eV. In consideration of the entropy effect of NO2 molecule, one can anticipate that this desorption process could occur at room temperature with a desorption free energy of 0.42 eV. Alternatively, the release of ONObri via the nitrate route (path Ⅲ in Scheme 2a) was also considered but seems to be less favorable. Specifically, ONObri could couple with another Obri nearby to form a bidentate NO3## species with a barrier of 0.91 eV and a reaction enthalpy of -0.53 eV; then, this NO3## could transform into a monodentate configuration endothermic by 0.20 eV, but the cleavage of the NO2-Obri bond to release NO2 into the gas phase is so difficult with a rather high barrier (Ea = 1.70 eV) and a large energy cost (ΔH = 1.51 eV). Accordingly, the overall reaction following path Ⅲ has an effective barrier as high as 1.37 eV (= 1.70 + 0.20 - 0.53 eV; Fig. S5 in Supporting information), and therefore is expected to be less favored relative to the direct desorption.
As for the formed Ovac, O2 could adsorb and trigger a series of possible reaction paths. It was found that O2 prefer to moderately adsorb at the Ovac site with the O-O bond parallel to the surface at an adsorption energy of -0.72 eV (Fig. S4c in Supporting information), in comparison with the upright adsorption configuration (Fig. S4d in Supporting information, Eads= -0.60 eV). Notably, either one of these two O2# configurations is difficult to dissociate at the Ovac with the barrier being above 1.31 eV, implying the reactant O2# also has to participate in the following reaction in the molecular state. Accordingly, three possible reaction pathways starting from O2# species were considered as illustrated by Scheme 2b, including the coupling reactions of O2# with NO* (path A), with the gaseous NO (path B) and with NO2# (path C), respectively.
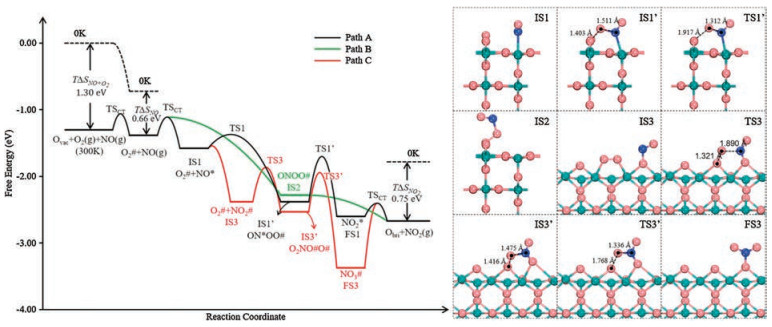
In path A, NO first adsorbs at the nearest Cr5c around the O2# with an adsorption energy of -0.86 eV, forming the co-adsorption configuration (IS1 in Fig. 2), which can quickly couple with each other into the ON*OO# species (IS1' in Fig. 2) without an evident barrier (Ea = 0.21 eV, and also being exothermic by 0.80 eV). Second, ON*OO# can overcome a barrier of 0.68 eV to break its O-O bond into NO2* and refill the Ovac simultaneously, corresponding to an enthalpy change of -0.22 eV; the optimized transition state (TS1') is shown in Fig. 2, and one can see that the O-O bond is elongated to 1.917 Å from the initial 1.403 Å in ON*OO#. Third, NO2* could desorb from Cr5c at ΔH = 0.82 eV and recovers the catalyst. The calculated free energy profile is illustrated in Fig. 2 (black line), and one can see that path A can efficiently occur without a significantly high barrier, revealing the feasibility of this path.

|
Download:
|
| Fig. 2. Left illustrated is the free energy profiles of NO oxidation on CrO2(110) for the refilling of the Ovac with O2# following three pathways: path A for the MvK reaction mechanism (black), path B for ER route (green) and path C for the nitrate route (red); notably, TSCT indicate the transition state estimated with the collision theory. Right are the optimized structures of some important intermediates and transition states of each path, and the key structural parameters are labeled. All the length values are in Å. | |
By comparison, in path B, the gaseous NO could directly couple with O2# to form ONOO# (IS2 in Fig. 2) without prior adsorption at the nearest Cr5c site, i.e., following the Eley-Rideal route. In term of total energy, this is a barrierless process and exothermic by -1.56 eV. Then, ONOO# can break its O-O bond to directly release NO2 into the gas phase at an energy cost of 0.50 eV. Comparing path B with A, which one is more favored depends on the quantitative kinetics when the overall reaction arrives at the steady state with the entropy effects included (see further the kinetic details).
Further, it is worth noting that O2# could also couple with NO2# to form a five-membered-ring intermediate O2NO#O# (IS3' in Fig. 2), corresponding to the nitrate formation/conversion route (path C in Scheme 2b). It requires a barrier of 0.52 eV, and the corresponding transition state (TS3) is shown in Fig. 2. Subsequently, the O-O bond cleavage in this O2NO#O# intermediate would refill the Ovac and lead to a monodentate NO3# species (FS3 in Fig. 2) with a barrier of 0.59 eV (TS3' in Fig. 2 for the corresponding TS). From the free energy profile (red line in Fig. 2), these two above steps can proceed readily; in particular, in comparison with the path A/B in Fig. 2, one can see that NO3# formation following path C (O2# +NO2# → O2NO#O# (IS3') → NO3# + O#) is even energetically more favored with a lower energy profile, implying its easy formation. However, the following NO3# dissociation to release NO2 has to surmount a large energy cost as high as 1.59 eV, limiting the overall activity. In other word, the Obri is too reactive to bind with NO2, resulting in the difficult removal of NO2 at room temperature, and the nitrate NO3# species could be gradually accumulated on CrO2(110) surface.
In addition, it is worth noting that NO could also adsorb at the Ovac site as a competitive adsorption species (NO# in Fig. S4e in Supporting information), which has a larger adsorption energy of -0.93 eV in comparison with that of O2 (-0.72 eV). Fortunately, it was found that NO# can couple with the neighboring Obri to form NO2#, which requires a low barrier of 0.25 eV and is exothermic by 0.85 eV. Therefore, NO# species may not result in serious poisoning effect to block the Ovac sites and inhibit O2 adsorption.
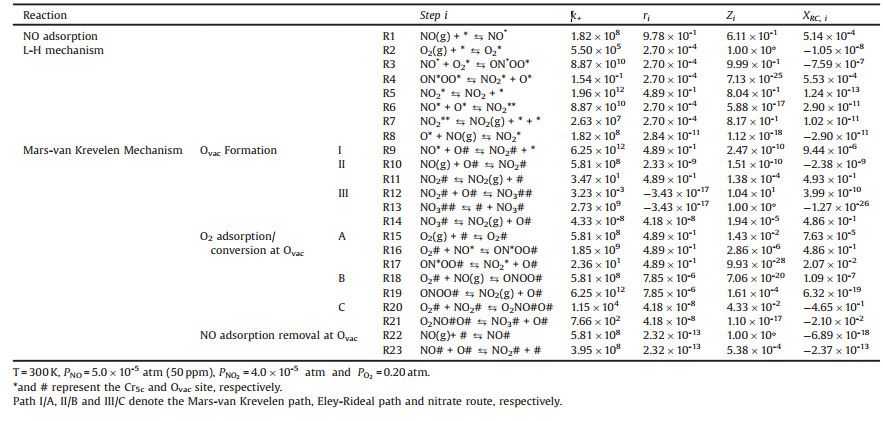
To figure out the favorable reaction mechanism of NO oxidation on CrO2(110) and quantitatively estimate the contribution/ poisoning effect of different reaction routes, the microkinetic analysis was carried out at room temperature (T = 300 K, PNO = 5.0 × 10-5 atm (50 ppm), and PO2 = 0.20 atm) (see details in the Supporting Information), in which all the above possible elementary steps were considered and the whole reaction network arrives at the steady state. The rate (ri) of each elementary step i were calculated in Table 1, and the surface coverage (θ(X)) of the intermediate species X on Cr5c or the lattice Obri sites (denoted as * and #, respectively) are shown in Table S1 (Supporting information). The following features can be obtained.
|
|
Table 1 Kinetic data, ri, Zi and XRC, i, for each elementary reaction step for the low-content NO oxidation over CrO2(110) at room temperature. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
First, with respect to the L-H mechanism (R1-R8) occurring on the Cr5c sites, NO can efficiently adsorb at Cr5c with a large coverage (θ(NO*) = 9.75×10-1), while the formation rate of O2* is small (2.70×10-4 s-1), according well with the weak adsorption of O2 and extremely low coverage of O2* (θ(O2*) = 4.85×10-12) on Cr5c. The corresponding contribution to NO2* formation is low. Thus, it quantitatively demonstrates that the L-H mechanism on Cr5c site alone (without the lattice Obri involved) contributes little to the activity of CrO2 for low-content NO oxidation at room temperature.
Second, the participation of surface lattice Obri is necessary to trigger the overall low-content NO oxidation reaction, and the Obri prefers to react with NO* to form NO2# (R9) rather than couple with the gaseous NO via the ER mechanism (R10) (4.89 ×10-1 vs. 2.33×10-9, s-1). For the release of the formed NO2#, at the steady state it is more likely to desorb as NO2 directly rather than to be converted through the nitrate route (4.89 ×10-1 vs. -3.43 ×10-17, s-1), i.e., NO2# first reacting with another Obri into the nitrate species and then releasing NO2 by breaking the O2N-O# bond. Besides, regarding the conversion of O2#, it also tends to readily couple with NO* via the MvK path. Therefore, one could realize that either the lattice Obri or O2# can easily oxidize NO*, which largely contributes the catalytic activity of CrO2(110) for NO oxidation at room temperature. Moreover, the Cr5c site and the lattice Obri site can efficiently adsorb NO and O2, respectively, guaranteeing the surface coverage of each species, and thus the synergetic role in between is critical for the overall activity of CrO2(110). For example, NO can predominately adsorb on the Cr5c sites (θ(NO*) = 9.75×10-1) over O2* (θ(O2*) = 4.85×10-12. Also, the high coverage of NO* eventually facilitates the O#/O2# to react with NO* instead of the gaseous NO with the ER route (4.89 ×10-1 vs. 2.33×10-9/ 7.85 ×10-6, s-1), despite that ER route is barrierless in the total energy as mentioned above.
Third, the coverage of the nirtrate NO3# species is very high (θ(NO3#) = 9.65×10-1), which results in the serious blocking effect for the lattice Ovac site and deactivates the CrO2(110) to some extent.
According to the above discussions, the favorable reaction pathways of low-content NO oxidation by CrO2(110) at room temperature (Figs. S2 and S3) can be summarized as follows. First, following the Mars-van Krevelen mechanism, the adsorbed NO* at Cr5c couples with the surface lattice Obri to form the ONObri species without an evident barrier, which desorbs as NO2 and form the lattice vacancy Ovac. Second, O2 adsorbs at Ovac and couples with NO*, leading to the intermediate ON*OO# spontaneously, which can break its O-O bond with a low barrier of 0.68 eV, yielding NO2* and refilling the Ovac. Third, the NO2* desorbs, completing the catalytic cycle. Fourth, the alternative coupling of O2# (the O2 adsorbed at Ovac) with NO2# would generate the stable nitrate species, which could hardly decompose and results in the surface accumulation (θ(NO3#) = 9.65 ×10-1), constituting the crucial poisoning effect toward the surface lattice Ovac sites. Nevertheless, following these reaction steps, the overall rate (turnover frequency, TOF) of room-temperature NO oxidation into NO2 on CrO2(110) is calculated to be as high as 9.78 × 10-1 s-1. This quantitatively demonstrates the high activity of CrO2 and that CrO2 could be one of the real active sites of CrOx-based catalysts. Interestingly, the very recent experimental result has uncovered the existence of CrO2 in efficiently promoting NO oxidation [26].
Furthermore, the "degree of rate control" (XRC, i, see details in Supporting Information) of each elementary step i was calculated in Table 1, aiming to quantitatively reveal the relative importance of each elementary step on affecting the overall catalytic activity. It can be found that the steps O2# + NO* 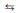
In summary, the reaction mechanism of low-content NO oxidation catalyzed by CrO2(110) surface at room temperature has been studied, by performing DFT calculations together with the microkinetic analysis, aiming to shed light on the catalytic ability of CrO2 and the inherent activity-limiting factors. The main result can be summarized as follows:
(ⅰ) CrO2(110) was revealed to have a potentially high activity in catalyzing NO oxidation at room temperature. Mechanically, the Langmuir-Hinshelwood path occurring at the Cr5c site alone is less active, while the overall reaction preferentially proceeds following the Mars-van Krevelen mechanism with the lattice Obri involved and the optimal reaction route is identified. Specifically, NO adsorbs at Cr5c and couples with the lattice Obri to form the ONObri species, which directly desorbs as NO2 and leaves an Ovac; then, O2 adsorbs at Ovac and couples with NO* into ON*OO# species, which can break its O-O bond with a low barrier to yield NO2* and refill Ovac; finally, the NO2* desorbs and completes the catalytic cycle.
(ⅱ) The nitrate NO3# species, mainly generated from the reaction of O2# with NO2#, was found to hardly decompose and results in the serious surface accumulation, which constitutes the crucial poisoning effect and deactivates the CrO2(110) catalyst. Originally, it indicates that the Obri on CrO2(110) is too reactive and bind NO2 tightly, and eventually limits the overall activity.
(ⅲ) Kinetically, the four steps, i.e., the formation of ON*OO#, NO2# desorption, NO3# formation and its decomposition, constitute the rate-limiting steps of NO oxidation network on CrO2(110).
This study extensively explored, at the atomic level, the catalytic ability of CrO2 for NO oxidation at room temperature, and the theoretical understanding may facilitate the further design of more active Cr-based catalyst.
AcknowledgmentsThis project was supported by the National Natural Science Foundation of China (NSFC, Nos. 21333003, 21622305), National Ten Thousand Talent Program for Young Top-notch Talents in China, The Shanghai Shuguang Scholar Program (No. 17SG30).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.12.018.
| [1] |
E. Fridell, M. Skoglundh, B. Westerberg, S. Johansson, G. Smedler, J. Catal. 183 (1999) 196-209. DOI:10.1006/jcat.1999.2415 |
| [2] |
Y. Sakamoto, K. Okumura, Y. Kizaki, et al., J. Catal. 238 (2006) 361-368. DOI:10.1016/j.jcat.2005.12.025 |
| [3] |
M.D. Amiridis, T. Zhang, R.J. Farrauto, Appl. Catal. B 10 (1996) 203-227. DOI:10.1016/0926-3373(96)00031-8 |
| [4] |
C. Ciardelli, I. Nova, E. Tronconi, D. Chatterjee, B. Bandl-Konrad, Chem. Commun. (2004) 2718-2719. |
| [5] |
Y. Mao, Z. Wang, H.F. Wang, P. Hu, ACS Catal. 6 (2016) 7882-7891. DOI:10.1021/acscatal.6b01449 |
| [6] |
J. Després, M. Elsener, M. Koebel, et al., Appl. Catal. B 50 (2004) 73-82. DOI:10.1016/j.apcatb.2003.12.020 |
| [7] |
D. Bhatia, R.W. McCabe, M.P. Harold, V. Balakotaiah, J. Catal. 266 (2009) 106-119. DOI:10.1016/j.jcat.2009.05.020 |
| [8] |
C. Wu, D.J. Schmidt, C. Wolverton, W.F. Schneider, J. Catal. 286 (2012) 88-94. DOI:10.1016/j.jcat.2011.10.020 |
| [9] |
J.M. Bray, W.F. Schneider, ACS Catal. 5 (2015) 1087-1099. DOI:10.1021/cs501783q |
| [10] |
C.H. Kim, G. Qi, K. Dahlberg, W. Li, Science 327 (2010) 1624-1627. DOI:10.1126/science.1184087 |
| [11] |
J.E. Parks, Science 327 (2010) 1584-1585. DOI:10.1126/science.1187154 |
| [12] |
W. Wang, G. McCool, N. Kapur, et al., Science 337 (2012) 832-835. DOI:10.1126/science.1225091 |
| [13] |
S.O. Choi, M. Penninger, C.H. Kim, W.F. Schneider, L.T. Thompson, ACS Catal. 3 (2013) 2719-2728. DOI:10.1021/cs400522r |
| [14] |
J.P.S. Sousa, M.F.R. Pereira, J.L. Figueiredo, Catal. Today 176 (2011) 383-387. DOI:10.1016/j.cattod.2010.11.040 |
| [15] |
Z. Zhang, J.D. Atkinson, B. Jiang, M.J. Rood, Z. Yan, Appl. Catal. B 148- 149 (2014) 573-581. |
| [16] |
B. Thirupathi, P.G. Smirniotis, Appl. Catal. B 110 (2011) 195-206. DOI:10.1016/j.apcatb.2011.09.001 |
| [17] |
Z. Chen, Q. Yang, H. Li, et al., J. Catal. 276 (2010) 56-65. DOI:10.1016/j.jcat.2010.08.016 |
| [18] |
H. Liu, L. Wei, R. Yue, Y. Chen, Catal. Commun. 11 (2010) 829-833. DOI:10.1016/j.catcom.2010.03.002 |
| [19] |
W. Cai, Q. Zhong, S. Zhang, W. Zhao, Chem. Eng. J. 236 (2014) 223-232. DOI:10.1016/j.cej.2013.09.032 |
| [20] |
W. Cai, Q. Zhong, S. Zhang, J. Zhang, RSC Adv. 3 (2013) 7009. DOI:10.1039/c3ra40226f |
| [21] |
W. Cai, Q. Zhong, W. Zhao, Chem. Eng. J. 246 (2014) 328-336. DOI:10.1016/j.cej.2014.02.090 |
| [22] |
S. Liu, M. Zhang, Y. Huang, et al., RSC Adv. 4 (2014) 29180-29186. DOI:10.1039/C4RA02681K |
| [23] |
L. Zhong, W. Cai, Y. Yu, Q. Zhong, Appl. Surf. Sci. 325 (2015) 52-63. DOI:10.1016/j.apsusc.2014.11.024 |
| [24] |
L. Zhong, W. Cai, Q. Zhong, RSC Adv. 4 (2014) 43529-43537. DOI:10.1039/C4RA05471G |
| [25] |
A. Wang, B. Lin, H. Zhang, et al., Catal. Sci. Technol. 7 (2017) 2362-2370. DOI:10.1039/C7CY00490G |
| [26] |
W. Cai, Q. Zhong, D. Wang, et al., ACS Appl. Nano Mater. 1 (2018) 1150-1163. DOI:10.1021/acsanm.7b00320 |
| [27] |
J. Jin, N. Sun, W. Hu, et al., ACS Catal. 8 (2018) 5415-5424. DOI:10.1021/acscatal.8b00081 |
| [28] |
G. Kresse, J. Furthmüller, Phys. Rev. B 54 (1996) 11169-11186. DOI:10.1103/PhysRevB.54.11169 |
| [29] |
G. Kresse, J. Furthmüller, Comput. Mater. Sci. 6 (1996) 15-50. DOI:10.1016/0927-0256(96)00008-0 |
| [30] |
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865-3868. DOI:10.1103/PhysRevLett.77.3865 |