 2019, Vol. 30
2019, Vol. 30 


b Fluorescence Research Group, Singapore University of Technology and Design, Singapore 487372, Singapore;
c University of Chinese Academy of Sciences, Beijing 100039, China
Organic fluorescent dyes have been widely used in various biological studies as fluorescent probes and labels, profiting from their small sizes, excellent photophysical and photochemical properties, and emissions spanning the entire color spectrum [1]. The fluorescence properties of fluorescent dyes are closely related to their molecular structures [2]. Therefore, a deep and systematic understanding of the luminescent structure-property relationship of dyes will enable more efficient and effective development of fluorescent dyes and fluorescent probes with improved properties. For example, different positions of substituents on fluorophores may significantly affect emission wavelengths and Stokes shifts [3]. Fluorescent properties are also strongly affected by environmental factors. Typically, with the increase of polarity, the fluorescence intensities of donor-acceptor type dyes decreases substantially with a bathochromic shift in both the UV-vis absorption and fluorescence spectra. Based on such environmental sensitivity, various fluorescent probes have been designed [4-8]. Besides, molecular aggregation plays a critical role in alternating the spectral characters of fluorophores. Currently, increasing research interests have been directed to investigate the impact of dye aggregation. For example, Tang et al. proposed the concept of aggregation-induced emission (AIE) [9]. Subsequently, numerous AIE molecules (or AIEgens) have been reported and received much attention [10]. Our groups also investigated the coumarin monomer-aggregate equilibrium and developed a temperature-insensitive fluorescent system [11].
Pyrene is an important class of fluorophores, broadly used in fluorescent labeling and sensing applications [12]. One of the most attractive features of pyrene dyes concerns their monomerexcimer switching, which can be used to study molecular interactions. 1-Acetylpyrene is a representative pyrene-derivative, used as a photoremovable protecting group [13], and an enzyme inhibitor [14]. Its fluorescence was believed to be strongly polaritysensitive (Fig. 1), which had been studied using absolute fluorescence quantum yield measurements and time-dependent density functional theory (TD-DFT) calculations [15]. These results showed that the fluorescence of 1-acetylpyrene was very weak in nonpolar solvents, but becomes quite intense in polar solvents. The fluorescence maximum displayed a significant red shift with increasing solvent polarity. This dye was subsequently applied to probe changes in environmental polarities [16]. Recently, it has been shown that 1-acetylpyrene is prone to molecular aggregation through solid-state structural studies [17]. The aggregation effect on fluorescence properties has also been investigated via varying dye concentrations [18]. For example, a Na+-selective podand-type receptor was developed by modulating the distance between two 1-acetylpyrene fluorophores, while the resulted emissions at 524 nm and 423 nm in chloroform have been ascribed to excimers and monomers, respectively [19]. In fact, the fluorescence between 410-470 nm, shown in Fig. 1b, has always been thought of monomer emissions of 1-acetylpyrene in different solvents.

|
Download:
|
| Fig. 1. (a) Chemical structures of compounds used in this study. (b) Fluorescence spectra of 1-acetylprene in different solvents (1 μmol/L). Excitation = 360 nm. (c) Fluorescence spectra of 1 before (dotted line) and after (solid line) absorbed on carbon nanotubes in water. | |
{kind=link}
However, several interesting findings caught our attention and led us to wonder if the monomer fluorescence of 1-acetylpyrene was indeed in the range of 410-470 nm and was highly polarity sensitive. Firstly, Paloniemi et al. discovered that 1 exhibited two fluorescence peaks in water (Figs. 1a and c), among which the short-wavelength peak was very similar to that of pyrenes [20]. Furthermore, when 1 was adsorbed onto the inside wall of carbon nanotubes, the long-wavelength peak disappeared. The remaining emissions, which centered at ~380 nm in aqueous solutions, seem to belong to the monomers of 1-acetylpyrene. Secondly, Ito et al. studied the fluorescence properties of nanoaggregates of ammonium-derived 1-acetylpyrene in tetrahydrofuran (THF)/aqueous solution [21]. Under continuous irradiations of 350 nm light, a new peak at 390 nm appeared at the expense of the other peak at 469 nm. This spectral change has been ascribed to the photochemical reactions between 1-acetylpyrene and THF, but without concrete experimental verifications. All these observations lead us to explore what emission peaks the monomers of 1-acetylpyrene possess, and whether monomer emission peaks are highly polarity sensitive.
In this paper, based on both experimental and theoretical studies, we discovered that the monomer fluorescence of 1-acetylpyrene is centered at 390 nm, which is very similar to that of pyrene. We showed that the monomer emissions are not much polarity-sensitive. We also found that fluorescence in the range of 410-470 nm is ascribed to dimers (and other molecular aggregates). Previous mis-assignment of dimer emissions to monomer emissions, is largely due to strong π-π stacking interactions between 1-acetylpyrene dyes, rending dimers and molecular aggregates as predominant forms in the solution phase.
In consideration of strong π-π stacking interactions between 1-acetylpyrene dyes, we decided to use host-guest chemistry to separate the dimers or aggregates, in order to reveal the true spectral properties of monomers. We thus synthesized compounds 2-4 (Fig. 1). Compound 2 contains an imidazolium group which can bind anions through electrostatic interactions. In particular, the imidazolium group in 2 is kept apart from the carbonyl group, minimizing possible intramolecular interactions. The neutral compound 4 contains a benzylsulfonamide ligand for binding with human carbonic anhydrase. Compound 3 was synthesized as a control.
We next investigated the UV-vis absorption and fluorescence properties of 2-3 in different solvents (Fig. 2). The peak UV-vis absorption wavelengths of 3 were much shorter than those of 2 (Figs. 2a and c). In various solvents, typical monomer and excimer dual emissions of pyrenes were observed in the solutions of 3 (Fig. 2d). In contrast, compound 2 demonstrated only one emission peak, which gradually shifts towards longer wavelengths as solvent polarity increases. These results were consistent with those reported in the literatures [15, 16]. When the concentration of 2 in aqueous solutions was diluted to 0.5 μmol/L, no obvious change was observed in the peak emission wavelength (Fig. S1 in Supporting information). At such a low concentration (5 × 10-7 mol/L), the aggregates or excimers were usually believed impossible to exist, and the observed emissions seem to be ascribed to monomers.

|
Download:
|
| Fig. 2. UV-vis and fluorescence spectra of compounds 2 (a-b) and 3 (c-d). Excitation = 360 nm. (e) Absorption spectral analysis of 2 treated with 1 mmol/L PPi. (f) Fluorescence spectra of 2 treated with different concentrations of PPi. Excitation = 345 nm. | |
{kind=link}
However, as we gradually added pyrophosphate (PPi) to the aqueous solution of 2, a new emission peak of < 400 nm gradually appeared (Fig. 1f). It is of note that the strong binding between PPi and imidazolium could dissolve the aggregate of 2, if any. The appearance of this new peak apparently suggests the existence of dimers/aggregates in the solution of 2. We tentatively assigned this new emission peak around 390 nm to the monomers of 2.
Given that the positive charge in imidazolium may affect the fluorescence properties of 2, we also synthesized a neutral compound 4, containing a side chain of benzylsulfonamide, which could specifically bind to carbonic anhydrase (Fig. 3). We expected that the binding of carbonic anhydrase with benzylsulfonamide (Fig. 3c) could dissolve the aggregate of 4. As shown in Fig. 3b, the addition of human carbonic anhydrase 1 (HCA1) induced a large increase of fluorescence intensities around 390 nm, indicating the generation of a new chemical spice, i.e., monomers. In addition, the fluorescence peak shifted from 460 nm to 440 nm.

|
Download:
|
| Fig. 3. (a) The mechanism of compound 4 binding with HCA 1 in PBS. After treated with the HCA 1 protein, 4 displayed monomer fluorescence due to the binding with the protein and the partial disassembly of molecular aggregates. (b) Fluorescence spectral analyses of 4 (1 μmol/L) in the absence (blue line) or presence (red line) of HCA 1 (1.3 μmol/L). Excitation = 345 nm. (c) Computer simulations of the protein binding with 4. | |
{kind=link}
It is of note that the dissociation constant between benzylsulfonamide and HCA1 was reported to be around 1 μmol/L [22]. The dissociation constant of imidazolium for PPi was estimated to be in the range of 1-100 μmol/L [12]. We speculated that the π-π stacking binding force between 1-acetylapyrene molecules is greater than 1 μmol/L. We're trying to use a stronger host and guest chemistry to separate the aggregates, such as the Avidinbiotin complex which is known to have the strongest non-covalent interaction (Kd = 10-15 mol/L), in order to further enhance the monomer emission signals.
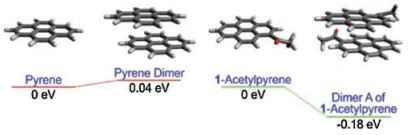
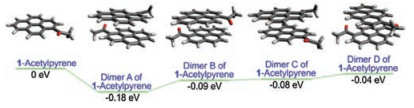
Next, we deployed computational chemistry to explore the π-π stacking interactions and optical properties of 1-acetylpyrene. Our computational results showed that 1-acetylpyrene has a stronger tendency to molecular aggregation, in comparison to pyrene. The relative Gibbs free energy of the dimer of 1-acetylpyrene is 0.18 eV more stable than that of its monomer, due to strong intermolecular π-π interactions. In contrast, the dimer of pyrene is 0.04 eV high than a pyrene monomer in the ground state (Fig. 4). During our calculations, we have considered four different conformations in the dimers of 1-acetylpyrene. Dimer A is the most stable one in the ground state (Fig. 5).

|
Download:
|
| Fig. 4. Comparison of the relative Gibbs free energy of pyrene, 1-acetylpyrene and their representative dimers in ethanol. | |
{kind=link}

|
Download:
|
| Fig. 5. Comparison of the relative Gibbs free energy of pyrene, 1-acetylpyrene and their representative dimers in ethanol. We have considered four different conformations in the dimers of 1-acetylpyrene. Dimer A is the most stable one. | |
{kind=link}
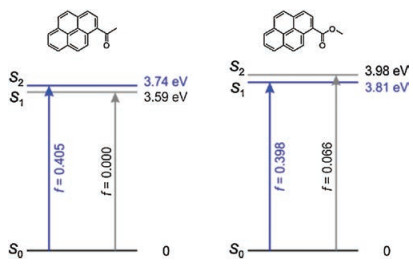
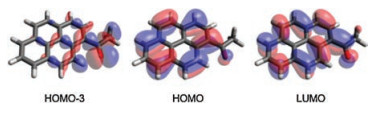
In addition, we noted that there exists a dark S1 state in 1-acetylpyrene (f = 0) in vacuo and weakly polar solvents, in contrast to a bright S1 state in 1-carboxylatepyrene (f = 0.398, Fig. 6). The dark S1 state in 1-acetylpyrene is mainly resulted from the optical transitions between HOMO-3 and LUMO (Fig. 7, Table S1 in Supporting information). As a result, the quantum yields of 1-acetylpyren are very low in most solvents.

|
Download:
|
| Fig. 6. Presence of a dark S1 state in 1-acetylpyren, in contrast to a bright S1 state in 1-carboxylatepyrene in vacuo. "f" denotes oscillator strength. | |
{kind=link}

|
Download:
|
| Fig. 7. HOMO-3, HOMO and LUMO of 1-acetylpyren (based on the optimized molecular structure of 1-acetylpyren in vacuo). | |
{kind=link}
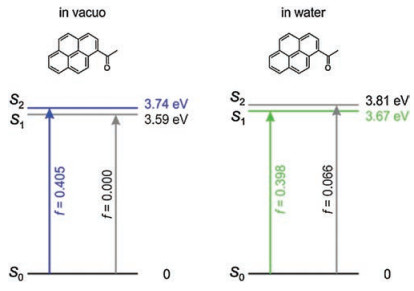
However, the bright state which mainly consists of HOMO-LUMO transition of 1-acetylpyrene becomes increasingly stable in polar protonic solvents. This transition relates to the S2 state in vacuo (f = 0.405) but becomes the S1 state in water (f = 0.398, Fig. 8). As the S1 state of 1-acetylpyrene become bright in water, we expected that the quantum yield of 1-acetylpyrene is high in water. These results are consistent with previously reported experimental data [15].

|
Download:
|
| Fig. 8. Energy levels of the S1 and S2 states of 1-acetylpyrene in vacuo (left) and water (right). | |
{kind=link}
Finally, our calculations show that the peak absorption (λabs) and emission (λem) wavelengths of 1-acetylpyrene and 1-carboxylatepyrene are not much sensitive to solvent polarities (Table S2 in Supporting information). As the solvent varies from dichloromethane to water, the variations in λabs and λem are within 2 nm and 20 nm, respectively. It is worth noting that there exist no significant differences between protonic (i.e., ethanol) and nonprotonic (i.e., acetonitrile) solvents. These results again support our claim that previously observed large solvatochromism of 1-acetylpyrene was due to the formation of dimers and molecular aggregates.
In conclusion, based on the experimental and theoretical studies, we showed the "true" monomer fluorescence of 1-acetylpyrene is centered at ~390 nm, which is very similar to that of pyrene and not polarity-sensitive. Fluorescence in the range of 410-470 nm is re-assigned to dimers and other forms of molecular aggregate. Interestingly, 1-acetylpyrene demonstrates strong intermolecular π-π interactions, rendering dimers and molecular aggregates as predominant forms in the solution phase. The lack of monomers led to the previous mis-assignment of dimer emissions to monomer emissions. Our results led to a deep understanding in the fluorescence properties of 1-acetylpyrene, and establish a foundation for the rationally deployment of this compound and derivatives in developing environmental sensitive probes. The dimer of 1-acetylpyrene with strong affinity may be used as a building block in supramolecular chemistry.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (Nos. 21878286, 21502189), DICP (Nos. DMTO201603, TMSR201601). WC and XL were indebted to the financial support from Singapore University of Technology and Design (SUTD) and the SUTD-MIT International Design Centre (Nos. T1SRCI17126, IDD21700101, IDG31800104). The authors would like to acknowledge the use of High-Performance Computing (HPC) service of both SUTD-MIT International Design Centre and National Supercomputing Centre (Singapore) in carrying out this work.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.12.008.
| [1] |
X. Qian, Z. Xu, Chem. Soc. Rev. 44 (2015) 4487-4493. DOI:10.1039/C4CS00292J |
| [2] |
X. Liu, Q. Qiao, W. Tian, et al., J. Am. Chem. Soc. 138 (2016) 6960-6963. DOI:10.1021/jacs.6b03924 |
| [3] |
(a) X. Liu, Z. Xu, J.M. Cole, J. Phys. Chem. C 117 (2013) 16584-16595; (b) X. Liu, J.M. Cole, Z. Xu, J. Phys. Chem. C 121 (2017) 13274-13279. |
| [4] |
(a) S. Leng, Q. Qiao, Y. Gao, et al., Chin. Chem. Lett. 28 (2017) 1911-1915; (b) L. Peng, Y. Xu, P. Zou, Chin. Chem. Lett. 28 (2017) 1925-1928; (c) Z. Xu, J. Chen, L. Hu, et al., Chin. Chem. Lett. 28 (2017) 1935-1942; (d) P. Ning, W. Wang, M. Chen, et al., Chin. Chem. Lett. 28 (2017) 1943-1951; (e) D. Wu, Y. Shen, J. Chen, et al., Chin. Chem. Lett. 28 (2017) 1979-1982. |
| [5] |
W. Chi, W. Yin, Q. Qi, et al., Mater. Chem. Front. 1 (2017) 2383-2390. DOI:10.1039/C7QM00345E |
| [6] |
Q. Qiao, W. Liu, J. Chen, et al., Dyes Pigm. 147 (2017) 327-333. DOI:10.1016/j.dyepig.2017.08.032 |
| [7] |
S. Leng, Q. Qiao, L. Miao, Chem. Commun. 53 (2017) 6448-6451. DOI:10.1039/C7CC01483J |
| [8] |
L. Dai, D. Wu, Q. Qiao, et al., Chem. Commun. 52 (2016) 2095-2098. DOI:10.1039/C5CC09403H |
| [9] |
J. Luo, Z. Xie, J.W.Y. Lam, et al., Chem. Commun. 37 (2001) 1740-1741. |
| [10] |
J. Mei, N.L.C. Leung, R.T.K. Kowk, et al., Chem. Rev. 115 (2015) 11718-11940. DOI:10.1021/acs.chemrev.5b00263 |
| [11] |
(a) X. Liu, D. Mao, J.M. Cole, et al., Chem. Commun. 50 (2014) 9329-9332; (b) D. Miao, X. Liu, Q. Qiao, et al., Analyst 14 (2015) 1008-1013. |
| [12] |
Z. Xu, N.J. Singh, J. Lim, et al., J. Am. Chem. Soc. 131 (2009) 15528-15533. DOI:10.1021/ja906855a |
| [13] |
(a) B.T. Tuten, J.P. Menzel, K. Pahnke, et al., Chem. Commun. 53 (2017) 4501-4504; (b) L. Pukenas, P. Prompinit, B. Nishitha, et al., ACS Appl. Mater. Interfaces 9 (2017) 18388-18397; (c) R. Kasprzyk, J. Kowalska, Z. Wieczorek, et al., Org. Biomol. Chem. 14 (2016) 3863-3868; (d) S. Barman, S.K. Mukhopadhyay, K.K. Behara, et al., ACS Appl. Mater. Interfaces 6 (2014) 7045-7054. |
| [14] |
T. Shimada, D. Kim, N. Murayama, et al., Chem. Res. Toxicol. 26 (2013) 517-528. DOI:10.1021/tx300492j |
| [15] |
Y. Niko, Y. Hiroshige, S. Kawauchi, et al., Tetrahedron 68 (2012) 6177-6185. DOI:10.1016/j.tet.2012.05.072 |
| [16] |
(a) A. Jana, S. Atta, S.K. Sarkar, et al., Tetrahedron 66 (2010) 9798-9807; (b) R.J. Berry, P. Douglas, M.S. Garley, et al., J. Photochem. Photobiol. A 120 (1999) 29-36; (c) K. Szczubiałka, Ł. Moczek, A. Goliszek, et al., J. Fluor Chem. 126 (2005) 1409-1418. |
| [17] |
(a) Y. Niko, S. Sasaki, K. Narushima, et al., J. Org. Chem. 80 (2015) 10794-10805; (b) S.K. Rajagopal, A.M. Philip, K. Nagarajan, et al., Chem. Commun. 50 (2014) 8644-8647. |
| [18] |
(a) F. Ito, T. Kakiuchi, T. Sakano, et al., Phys. Chem. Chem. Phys. 12 (2010) 10923-10927; (b) R. Flamhol, D. Plazuk, J. Zakrzewski, et al., RSC Adv. 4 (2014) 31594_-31601. |
| [19] |
Y. Nishimura, T. Takemura, S. Aril, ARKIVOC x (2009) 43-52. |
| [20] |
H. Paloniemi, T. Ääritalo, T. Laiho, et al., J. Phys. Chem. B 109 (2005) 8634-8642. DOI:10.1021/jp0443097 |
| [21] |
F. Ito, T. Sagawa, H. Koshiyama, Res. Chem. Intermediat. 41 (2015) 6897-6906. DOI:10.1007/s11164-014-1786-3 |
| [22] |
C.T. Supuran, Nat. Rev. Drug Discov. 7 (2008) 168-181. DOI:10.1038/nrd2467 |