 2019, Vol. 30
2019, Vol. 30 


The asymmetric conjugate addition of stabilized carbanions to Michael acceptors is one of the fundamental C-C bond forming reactions in organic chemistry [1]. The research of the asymmetric version of this reaction constitutes an important research field and has been extensively explored in recent years [2]. Among various types of Michael donors, nitro esters are valuable sources of stable carbanions in organic synthesis. Additionally, nitro esters can be converted into many biologically active compounds [3]. Therefore, the asymmetric Michael reactions employing nitroacetates as donors have been an attractive study to the synthetic chemists [4]. In 2006, Wang's group studied an organocatalytic enantioselective conjugate addition of nitro esters catalyzed by a cinchona alkaloid [5a]. In 2010, Lu's group developed an enantioselective conjugate addition of nitro esters to α, β-unsaturated ketones with an amine-based organocatalyst [5b]. After that, an enantioselective conjugate addition of nitro esters to β, γ-unsaturated α-ketoesters with a chiral copper catalyst was achieved by our group [6]. Meanwhile, 2-enoylpyridine N-oxide, which is also one of the most popular substrates, have been well-explored in asymmetric transformations due to the good coordination ability of pyridine N-oxide [7]. Our group also have been focusing on this enantioselective conjugate addition for a long time [6, 8]. However, to the best of our knowledge, the 1, 4-addition of nitro esters to 2-enoyl-pyridine N-oxides has not been explored yet. In this context, we report our recent efforts in developing an enantioselective Michael addition of nitro esters to 2-pyridine N-oxide (Scheme 1).

|
Download:
|
| Scheme 1. Enantioselective Michael addition of nitro esters to 2-enoyl-pyridine N-oxides. | |
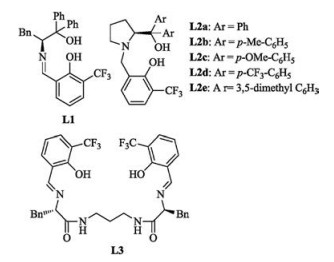
Our investigation began with the model reaction of 2-enoylpyridine N-oxide 1a with ethyl nitroacetate 2a (Table 1). At first, the Shiff base ligand L1 (Fig. 1) was employed in this reaction (entry 1). However, the desired product 3aa was not detected. When the C2-symmetric Schiff-base ligand L3 was examined, only a trace amount of product 3aa was detected (entry 7). Then, different tridentate ligands were examined, as shown in Table 2. To our delight, the chiral ligand L2a was able to catalyze this reaction with good enantioselectivity (entry 2).
|
|
Table 1 Optimization of the reaction.a |


|
Download:
|
| Fig. 1. Structures of ligands L1-L3. | |
Therefore L2 was chosen as a ligand in this reaction. It was found that L2 worked well in this reaction (Table 1, entries 2–6) and the L2a was the optimal ligand for this reaction (entry 2). Subsequently, the solvent optimization was finished and toluene proved to be the best solvent for this reaction, as shown in Table S1 in supporting information. Then the reaction temperature optimization showed that 0 ℃ should be the best reaction temperature, seeing the Table S1. Various metal salts were screened in this reaction. The optimization of the metal salts showed that Cu(OAc)2·H2O proved to be the optimal metal salt (entry 9). To further optimize the reaction conditions, increasing the amount of volume of toluene to 2.5 mL led to the formation of target product 3aa in 95% yield with 93/94 (%) ee (entry 14). Finally, the optimal conditions were determined as described below: Cu (OAc)2·H2O was employed as the copper salt, L2a as chiral ligand and 2 mL of toluene as the solvent.
With the optimal conditions in hand, the substrate scope of 2- enoyl-pyridine N-oxide 1 was studied (Table 2). Firstly, the electronic effect of the R1 in substrates was investigated. When R1 was the substituted phenyl group, it was found that the R1 with electron-donating groups on the para-position of the phenyl ring gave the desired products with high yields and good enantioselectivities (entries 2, 3). Meanwhile, when the R1 bore weak electron-withdrawing groups, such as halogens, or strong electron-withdrawing, such as trifluoromethyl group, the reaction could also be carried out smoothly to give the desired products with high yields and excellent enantioselectivities (entries 4–7). This indicated that electronic effect had little influence on this reaction.
|
|
Table 2 Conjugate addition of nitroacetates to 2-enoyl-pyridine N-oxides.a |

{kind=link}
{kind=link}
Then, the position of the substituents on the phenyl ring of R1 was examined. It was found that the yield of 3ha, 3ja, which bearing ortho substituent, was slightly lower than those of 3ba, 3ea, which bearing para substituent, and 3ia, 3ka, which bearing meta substituent (entries 2, 5, 8–11). This implied that steric effect on this phenyl ring disfavored the reaction yield but had little influence on the stereoselectivity. The R1 substituted phenyl group can be varied in this reaction. For instance, when R1 was 2- naphthyl group, the reaction can be carried out smoothly to give the corresponding product 3ma with high yield and excellent enantioselectivity. Similarly, steric effect was observed for this naphthyl R1. When R1 was 1-naphthyl group, for example, the product 3la can be obtained with a slightly lower yield in comparison with the product 3ma (entries 12, 13). In the case of R1 being heterocyclic group, the desired product could also be obtained in the moderate yields and good to high enantioselectivities (entries 14, 15). Moreover, when the R1 group was changed to the aliphatic group, the desired product 3pa was still obtained in good yield and excellent enantioselectivity (entry 16). Subsequently, the R2 effect on the reaction was investigated. When R2 was changed into tert-butyl group from ethyl group, the corresponding product 3ab could be obtained with a good yield of 84% and a slightly lower ee value of 90/90% with a longer reaction time of 48 h, perhaps due to the hindrance from the large volume of tert-butyl group (entry 17).
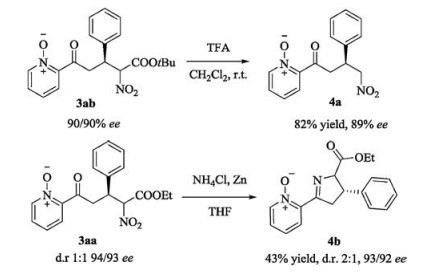
The synthetic utility of this asymmetric transformation was demonstrated by the conversion of the reaction adducts into 4a, as shown in Scheme 2. The reaction adduct 3ab underwent a facile acidic hydrolysis to remove an ester group, followed by in situ decarboxylation, affording (S)-2-(4-nitro-3-phenylbutanoyl)-pyridine N-oxide (4a) in a good yield and with a high enantioselectivity [8]. This could be a practical method to produce asymmetric Michael addition products of nitromethane to 2-enoyl-pyridine N-oxides. Moreover, an analogue of nicotine, dihydro-2H-pyrrol 4b was synthesized as another synthetic utility of this asymmetric transformation (Scheme 2) [9]. The adduct 3aa underwent a facile reduction procedure which included the reduction of the nitro group and in situ cyclization, affording biologically active dihydro- 2H-pyrrol 4b in a moderate yield and with a high enantioselectivity. Because of different reaction activation of two diastereoisomers of 3aa, the corresponding product 4b was obtained with a 2:1 diastereoselectivity.

|
Download:
|
| Scheme 2. Synthetic manipulation of the Michael addition products. | |
{kind=link}
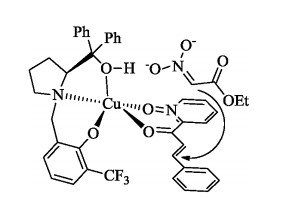
On the basis of our previous mechanism study [6, 8], a plausible structure of the transition state was proposed as shown in Fig. 2.

|
Download:
|
| Fig. 2. A plausible structure of the transition state of Michael addition of 2-enoylpyridine N-oxide with ethyl nitroacetate. | |
{kind=link}
In summary, a highly enantioselective copper-catalyzed Michael addition of 2-enoyl-pyridine N-oxide with nitro acetates was first developed under mild conditions. A series of chiral adducts were obtained with excellent enantioselectivities and good yields. Asymmetric Michael addition products of nitromethane to 2-enoyl-pyridine N-oxide can be produced in one step. An analogue of nicotine, dihydro-2H-pyrrol 4b was synthesized. Moreover, the plausible structure of transition-state was proposed.
AcknowledgmentsWe are grateful to the financial support from the National Nature Science Foundation of China (Nos. 21432009, 21672200, 21472177, 21772185) and the assistance of the product characterization from the Chemistry Experiment Teaching Center of University of Science and Technology of China. This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB20000000).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j. cclet.2018.11.024.
| [1] |
(a) G. Bartoli, Acc. Chem. Res. 17 (1984) 109-115; (b) R. Ballini, G. Bosica, D. Fiorini, A. Palmier, M. Petrini, Chem. Rev. 105 (2005) 933-971. |
| [2] |
(a) M.P. Sibi, S. Manyem, Tetrahedron 56 (2000) 8033-8206; (b) O.M. Berner, L. Tedechi, D. Enders, Eur. J. Org. Chem. (2002) 1877-1894; (c) J. Christoffers, A. Baro, Angew. Chem. 115 (2003) 1726-1728 Angew. Chem. Int. Ed. 42 (2003) 1688-1690; (d) W. Notz, F. Tanan ka, C.F. Barbas Ⅲ, Acc. Chem. Res. 37 (2004) 580-591; (e) A. Alexakis, J.E. Bäckvall, N. Krause, O. Pãmies, M. Diéguez, Chem. Rev. 108 (2008) 2796-2823; (f) X. Yang, X. Zhou, L. Lin, et al., Angew. Chem. Int. Ed. 47 (2008) 7079-7081; (g) T. Jerphagnon, M.G. Pizzuti, A.J. Minnaard, B.L. Feringa, Chem. Soc. Rev. 38 (2009) 1039-1075; (h) M. Hua, H. Cui, L. Wang, J. Nie, J. Ma, Angew. Chem. Int. Ed. 49 (2010) 2772-2776; (i) L. Wang, Q. Zhang, X. Liu, et al., Chem. Eur. J. 16 (2010) 7696-7699; (j) Z. Wang, D. Chen, Z. Yang, et al., Chem. Eur. J. 16 (2010) 10130-10136. |
| [3] |
(a) B.M. Trost, J.J. Surivet, J. Am. Chem. Soc. 122 (2000) 6291-6292; (b) B.M. Trost, J.J. Surivet, Angew. Chem. 112 (2000) 3252-3254 Angew. Chem. Int. Ed. 39 (2000) 3122-3124; (c) B. Moreau, A.B. Charette, J. Am. Chem. Soc. 127 (2005) 18014-18015; (d) A. Singh, R.A. Yoder, B. Shen, J.N. Johnston, J. Am. Chem. Soc. 129 (2007) 3466-3467; (e) A.D. Young, M.C. White, J. Am. Chem. Soc. 130 (2008) 14090-14091; (f) M. Aginagalde, T. Bello, C. Masdeu, et al., J. Org. Chem. 75 (2010) 7435-7438. |
| [4] |
(a) N. Halland, R.G. Hazell, K.A. Jørgensen, J. Org. Chem. 67 (2002) 8331-8338; (b) T. Ikariya, H. Wang, M. Watanabe, K. Murata, J. Organomet. Chem. 689 (2004) 1377-1381; (c) H. Li, Y. Wang, L. Tang, et al., Angew. Chem.117 (2005) 107-110 Angew. Chem. Int. Ed. 44 (2005) 105-108; (d) A. Prieto, N. Halland, K.A. Jørgensen, Org. Lett. 7 (2005) 3897-3900; (e) J. Wang, H. Li, L. Zu, et al., J. Am. Chem. Soc. 128 (2006) 12652-12653; (f) H. Lu, X. Wang, C. Yao, et al., Chem. Commun. (2009) 4251-4253; (g) X. Li, B. Wang, J. Zhang, M. Yan, Org. Lett. 13 (2011) 374-377; (h) A. Quintard, A. Alexakis, Org. Biomol. Chem. 9 (2011) 1407-1418; (i) S. Shirakawa, S.J. Terao, R. He, K. Maruoka, Chem. Commun. 47 (2011) 10557-10559; (j) R. Lu, W. Wei, J. Wang, et al., Tetrahedron 68 (2012) 9397-9404. |
| [5] |
(a) J. Wang, H. Li, L. Zu, et al., J. Am. Chem. Soc. 128 (2006) 12652-12653; (b) C. Liu, Y. Lu, Org. Lett. 12 (2010) 2278-2281. |
| [6] |
S. Zhang, K. Xu, F. Guo, et al., Chem. Eur. J. 20 (2014) 979-982. DOI:10.1002/chem.201303512 |
| [7] |
(a) A. Landa, A. Minkkil, G. Blay, K. Jørgensen, Chem.-Eur. J. 12 (2006) 3472-3483; (b) A. Landa, B. Richter, R. Johansen, A. Minkkil, K. Jørgensen, J. Org. Chem. 72 (2007) 240-245; (c) S. Barroso, G. Blay, J. Pedro, Org. Lett. 9 (2007) 1983-1986; (d) P. Singh, V. Singh, Org. Lett. 10 (2009) 4121-4124; (e) P. Singh, V. Singh, Org. Lett. 12 (2010) 80-83; (f) S. Barroso, G. Blay, M. Muñoz, J. Pedro, Org. Lett. 13 (2011) 402-405; (g) S. Ray, P. Singh, V. Singh, Org. Lett. 13 (2011) 5812-5813; (h) A. Livieri, M. Boiocchi, G. Desimoni, G. Faita, Chem.-Eur. J. 18 (2012) 11662-11668; (i) S. Ray, P. Singh, N. Molleti, V. Singh, J. Org. Chem. 77 (2012) 8802-8808; (j) S. Ray, S. Rout, V. Singh, Org. Biomol. Chem. 11 (2013) 2412-2416; (k) J. George, B. Subba Reddy, Adv. Synth. Catal. 355 (2013) 383-388; (l) M. Holmquist, G. Blay, M. Muñoz, J. Pedro, Org. Lett. 16 (2014) 1204-1207; (m) G. Desimoni, G. Faita, P. Quadrelli, Chem. Rev. 114 (2014) 6081-6129; (n) Y. Lu, Y. Zhou, L. Lin, et al., Chem. Commun. 52 (2016) 8255-8258. |
| [8] |
L. Li, S. Zhang, Y. Hu, et al., Chem.-Eur. J. 21 (2015) 12885-12888. DOI:10.1002/chem.201502129 |
| [9] |
(a) I.A. McDonald, N. Cosford, J.M. Vernier, Annu. Rep. Med. Chem. 30 (1995) 41-50; (b) D. Pogocki, T. Ruman, M. Danilczuk, et al., Eur. J. Pharmacol. 563 (2007) 18-39; (c) R. Srinivasan, B.J. Henderson, H.A. Lester, C.I. Richards, Pharmacol. Res. 83 (2014) 20-29. |