 2019, Vol. 30
2019, Vol. 30 


b Institute of Interdisciplinary Integrative Medicine Research, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China;
c Biotechnology Department, College of Basic Medical Sciences, Dalian Medical University, Dalian 116044, China;
d School of Life Science and Medicine, Dalian University of Technology, Panjin 124221, China
Carboxylesterase 1 (CES1), one of the most abundant serine hydrolases in mammals, involves in many physiological processes via hydrolysis of variety of endogenous esters and xenobiotic esters [1-3]. Recent studied have demonstrated that CES1 inhibitors can be used to modulate endogenous metabolism or to improve the outcomes of patients administrated ester drugs [4-6]. Thus, the tools for deciphering the relevance of CES1 to human diseases and for high throughput screening (HTS) of CES1 inhibitors are highly desirable. Unfortunately, the physiologically relevant substrates (such as cholesterol esters) for CES1 display poor specificity, low detection sensitivity and incapable for HTS of CES1 inhibitors [7, 8]. Thus, development of practical and reliable assays for rapid, sensitive and real-time monitoring CES1 activities in complicated biological systems is urgently needed, which can strongly facilitate CES1-related drug discovery, clinical practice and the basic researched on CES1-associated physiological or pathological processes.
Over the past decade, fluorescent probes have been widely used for sensing bio-molecular due to their inherent advantages including ultrahigh sensitivity, easy management and real-time response toward target analytes [9-15]. However, until now, the practical and reliable fluorescent probes for highly selective sensing CES1 in biological systems are seldom reported, due to the following two challenges. Firstly, the overwhelming majority of fluorophores are polycyclic phenolic compounds and their corresponding esters are more likely to be hydrolyzed by CES2. This explains why many isoform-specific fluorescent probes for CES2 or the fluorescent co-substrates of CES have been reported, while the isoform-specific fluorescent probes for CES1 are rarely reported [16-21]. Secondly, there are many serine hydrolases (such as proteases, peptidases and lipases) distributed in the human body, while all known serine hydrolases contain a conserved catalytic triad [22]. The conserved serine catalytic triad makes the substrate spectra of mammalian serine hydrolases are highly overlapped, which is a big challenge for constructing the isoformspecific probes for CES1 [23]. Four years ago, we have reported a highly specific fluorescent probe (termed BMBT) for CES1 [24]. However, the short emission wavelength of BMBT (< 500 nm) strongly limits its applications in complicated biological systems [25]. Thus, it is necessary to develop more practical fluorescent probes for highly selective sensing CES1 activities in various biological systems, as well as for HTS of CES1 inhibitors using unpurified enzyme as enzyme source.
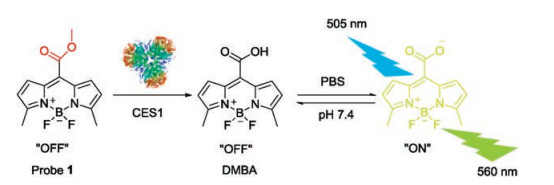
Herein, 3, 5-dimethyl BODIPY acid (DMBA) was used as the basic skeleton due to the good features of BODIPY dyes including high fluorescence quantum yield, good biocompatibility, high chemical stability and photochemical stability, and easy to chemical modification [26-28]. Furthermore, DMBA contained a large carboxyl part, its ester derivatives with small alcohol groups might be the good substrate for CES1 [29]. In this case, a number of ester derivatives of DMBA were rationally synthesized on the basis of substrate preference of CES1 (Fig. S1 in Supporting information). Preliminary study showed that the methyl ester of DMBA (probe 1) displayed excellent specificity (Fig. S2 in Supporting information), high sensitivity and fast respond towards CES1 over other esterases [30], which encouraged us to fully characterize the performance and applicability of probe 1 for sensing CES1 activities in complicated biological systems.
Firstly, the stability of probe 1 was investigated under physiological conditions (pH 7.4, 37 ℃). As shown in Figs. S3-S6 (Supporting information), probe 1 was very stable under physiological conditions (pH 7.4, 37 ℃). No hydrolytic metabolite was detected following 60 min incubation in all tested systems without CES1. By contrast, upon addition of CES1 or CES1- containing samples, probe 1 could be rapidly converted to DMBA and bring strongly fluorescent signal around 560 nm (Scheme 1). Then, both reaction phenotyping assays and chemical inhibition assays were used to validate the specificity of probe 1 toward CES1. probe 1 can be selectively hydrolyzed by CES1 while other serine hydrolyses displayed negligible effects toward probe 1 (Fig. S7A in Supporting information). More importantly, BNPP (bis-p-nitrophenyl phosphate, a CES inactivator) and UKA (3-O-(β-carboxypropionyl)-ursolic acid, a selective inhibitor of CES1) could significantly inhibit probe 1 hydrolysis in both recombinant human CES1 and HLMs, while GA (galanthamine, a specific inhibitor of AChE), LPA (loperamide, a specific inhibitor of CES2) and EDTA (ethylene diamine tetraacetic acid, a specific inhibitor of PONs) exhibited very weak inhibitory effects on probe 1 hydrolysis (Fig. S7B in Supporting information). These results suggested that probe 1 could be a specific probe for sensing the real activities of CES1 in complicated biological samples.

|
Download:
|
| Scheme 1. The schematic illustration of probe 1 for detection of CES1 in biological system. | |
{kind=link}
Next, the enzymatic kinetic of probe 1 hydrolysis was carefully investigated, owing to it is very important for quantitative applications of probe substrates [31-33]. As shown in Fig. S8 (Supporting information), probe 1 hydrolysis in both recombinant human CES1 and HLMs followed classic Michaelis-Menten kinetics. The kinetic parameters (Km and Vmax) of probe 1 hydrolysis in both CES1 and HLMs were determined and listed in Table S1 (Supporting information). The Km value of probe 1 hydrolysis in CES1 was much closed to that in HLMs, suggesting that CES1 was the major enzyme in HLMs responsible for probe 1 hydrolysis. Furthermore, probe 1 exhibited high affinity (km < 5 μmol/L) and satisfied reactivity (kcat/km > 5 mL min-1 mg-1) towards target enzyme CES1. These results clearly demonstrated that probe 1 displayed ideal kinetic behavior and satisfied reactivity for sensing human CES1 in biological samples, which encouraged us to quantify CES1 in human samples by using probe 1 as a probe substrate.
Encouraged by the excellent specificity and ideal kinetic behavior of probe 1 toward CES1, we subsequently use probe 1 to quantify CES1 in biological samples. Prior to quantification assays, the linear response and the detection sensitivity of probe 1 towards CES1 were investigated. As shown in Fig. 1a, a good linearity (R2 = 0.9833, P < 0.001) between fluorescence intensities of DMBA (560 nm) and incubation time (less than 40 min) were observed. As shown in Fig. 1b, an excellent linear correlation (R2 = 0.9967, P < 0.001) between the fluorescence intensities at 560 nm and CES1 concentrations ranging from 0 to 2.0 mg/mL, was acquired under physiological conditions (pH 7.4 at 37 ℃ for 30 min). We subsequently used probe 1 to quantify the CES1 activities in real samples, including a panel of human tissue preparations from individuals. As shown in Fig. 1c, the enzymatic activities of CES1 from various tissues including liver, lung, intestine and kidney were assayed, in which HLMs displayed the highest CES1 activities while CES1 activities in intestine and kidney were extremely low. Notably, these results were highly consistent with the protein levels of CES1 determined by western blotting (Fig. 1d). Furthermore, we also assayed the hydrolytic activities of CES1 in a panel of HLMs using both probe 1 and clopidogrel (a known CES1 substrate drug) as probe substrates for CES1. As shown in Fig. 1e, about 5-fold difference in CES1 activities among these individual samples were observed, which agreed well with previous literatures regarding the variability in both expression and activity of CES1 in HLMs [34-36]. Moreover, A high correlation coefficient (R2 = 0.9549, P < 0.001) between the hydrolytic rates of probe 1 and DMBA were observed (Fig. 1f). These results clearly suggested that probe 1 hydrolysis could work as a probe reaction for sensing human CES1 activities in real biological samples.

|
Download:
|
| Fig. 1. (a) Time-fluorescence responses of probe 1 (10 μmol/L) after incubated with recombinant human CES1 (1 mg/mL) and (b) enzyme concentration-fluorescence responses of probe 1 (10 μmol/L) following incubation with recombinant human CES1 (0.1–2 mg/mL) at 37 ℃ for 30 min (λex/λem = 505/560 nm). (c) Comparison of the hydrolytic rates and expression levels of human CES1 in different tissues microsomes, (d) western blotting analysis of human CES1 expression levels in different microsomes. (e) The hydrolytic rates of probe 1 in a number of individual HLMs (n = 12), and (f) correlation analysis between the hydrolytic rates of probe 1 and clopidogrel in these individual HLMs (n = 12). | |
{kind=link}
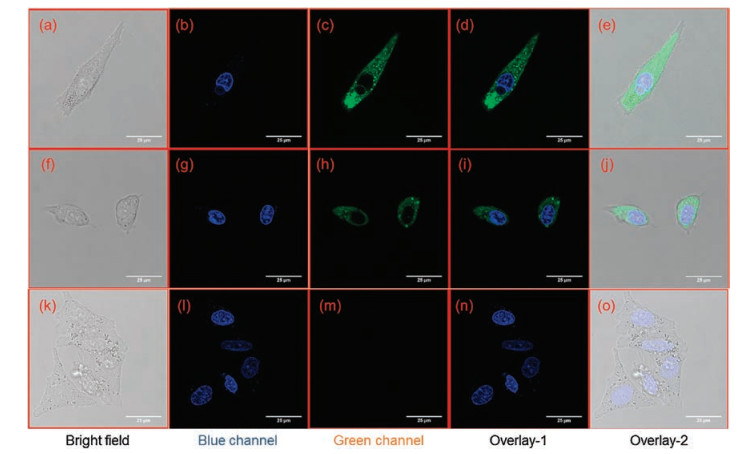
The above findings encouraged us to use probe 1 as a promising tool for real-time monitoring CES1 activities in living systems. Prior to cell imaging, the cytotoxicity of probe 1 and its hydrolytic metabolite (DMBA) were assayed by CCK-8 assays. As shown in Fig. S9 (Supporting information), both probe 1 and DMBA displayed weak cytotoxicity to A549 cells, and more than 80% cell viability of A549 cells were observed following co-incubation with probe 1 (80 μmol/L) or DMBA (80 μmol/L) for 2 h at 37 ℃. After that, probe 1 was used to monitor intracellular CES1 in A549 cells, while the confocal image results were recorded in both blue (for Hoechst 33342) and green (for DMBA) channels. As shown in Figs. 2a-e, after staining with probe 1 (2 μmol/L) at 37 ℃ for 30 min, A549 cells displayed strong green fluorescence signals (for DMBA) with excitation wavelength at 488 nm. In contrast, the fluorescence signals in the green channel of A549 cells pre-treated with BNPP could be strongly blocked (Figs. 2f–j). To further validate that probe 1 hydrolysis in A549 cells was catalyzed by CES1, chemical inhibition assays were conducted in A549 cell lysates. These results demonstrated that probe 1 hydrolysis in A549 cell lysates could be remarkably reduced by BNPP, while LPA could not inhibit probe 1 hydrolysis in A549 cell lysates (Fig. S10 in Supporting information). These findings suggested that probe 1 was cell membrane permeable and could work as a practical tool for sensing CES1 activities in living cells, which would be very helpful for deciphering CES1-associated physiological functions and its relevance to human diseases in living systems.

|
Download:
|
| Fig. 2. Imaging (63× magnification) of endogenous CES1 in A549 cells using probe 1 as a fluorescent substrate. (a–e) Cells stained with probe 1 (2μmol/L) and Hoechst 33342 for 30 min; (f–j) Cells pre-incubated with BNPP (100 μmol/L) for 30 min and then stained with probe 1 (2 μmol/L) and Hoechst 33342 for another 30 min; (k–o) Cells only incubated with Hoechst 33342 for 30 min (the concentration of Hoechst 33342 was used according to instruction, blue channel: 440–480 nm, green channel: 540–580 nm, scale bar = 25 μm). | |
{kind=link}
To further assess the applicability of probe 1 as a practical fluorescent probe for imaging of CES1 ex vivo or in vivo, its sensing ability for CES1 in mouse liver slices and zebrafish were investigated. Prior to tissue imaging, we investigated the specificity of probe 1 toward Ces3 in mouse (the ortholog of human CES1). As shown in Fig. S11 (Supporting information), probe 1 hydrolysis in mouse liver preparations could be strongly inhibited by BNPP, while LPA displayed negligible effects on probe 1 hydrolysis in mouse liver preparations. This finding suggested that the hydrolysis of probe 1 in mouse liver is mouse Ces3 dependent. In this case, mouse liver slice was incubated with probe 1 in PBS (pH 7.4, 37 ℃) for 30 min and subsequently washed three times with PBS to remove the residual substrate. As shown in Fig. S12 (Supporting information), under excitation at 488 nm, a strong fluorescence signals at green channel (540–580 nm) was observed and the depth of tissue penetration was as far as 40 μm. These findings demonstrated that probe 1 was capable of measuring endogenous Ces in living tissues by confocal microscope, which may attributed to the excellent optical properties of BODIPY dyes [26].
As a commonly used vertebrate model in developmental biology, toxicology and health related fields [37], zebrafish was also a preferred model for fluorescence imaging of target analytes. Notably, CES was also abundant in zebrafish, while Ces3 in zebrafish was the ortholog of human CES1 [38]. As shown in Fig. 3, after co-incubation with probe 1 for 30 min, the liver displayed strongly fluorescence at green channel, while no background signals or auto-fluorescence phenomenon were observed in zebrafish. This result was in accordance with a previous literature that Ces3 was highly expressed in liver of zebrafish [39]. By contrast, upon addition of BNPP, the fluorescence intensity at green channel (500–600 nm) was significantly reduced, suggesting that probe 1 hydrolysis in zebrafish was also Ces-dependent. To the best of our knowledge, it was the first time to realize in situ imaging of CES1 activities in zebrafish. These findings demonstrated that probe 1 could be used for sensing endogenous CES1 in living tissues and zebrafish, which held great promise for further investigations on CES1-associated physiological or pathological processes in living animals.

|
Download:
|
| Fig. 3. Biological imaging (4× magnification) of Ces activity in zebrafish. (a–c) Zebrafish incubated with probe 1 (2 μmol/L) for 30 min, (d–f) zebrafish preincubated with BNPP (100 μmol/L) for 30 min, then incubated with probe 1 (2 μmol/L) for another 30 min, (g–i) zebrafish only, scale bar = 500 μm. L. Ding et al. / Chinese Chemical Letters 30 (2019) 558–562 561 μm). | |
{kind=link}
As mentioned above, a reliable and practical assay for HTS of CES1 inhibitors would be great helpful for the discovery of drug candidates to treat CES1-associated metabolic disorders, such as hypertriglyceridemia and obesity [4, 40]. It should be noted that CES1 is an intracellular target which localized within the lumen of the endoplasmic reticulum. Thus, a practical cell-based assay for screening inhibitors targeting intracellular CES1 was necessary. Taking into account that probe 1 displayed good cell permeability and low cytotoxicity, as well as the good optical properties of DMBA and the relatively high expression of CES1 in HepG-2 cells, probe 1 was used to screen CES1 inhibitors in living HepG-2 cells directly. To demonstrate the potential use of probe 1, a fluorescence-based inhibition assay for screening of CES1 inhibitors was established using the fluorescence microplate reader as the detection system. As show in Fig. 4, both BNPP and UKA could strongly inhibit the catalytic activities of CES1 in recombinant human CES1 and HLM via a dose-dependent manner, but the IC50 values of these two inhibitors were much higher than that in recombinant human CES1 or in HLM. The IC50 value of BNPP on cellular CES1 in HepG-2 cells was evaluated as 854 nmol/L (Table S2 in Supporting information), which was about 8-fold of that in HLM. The IC50 value of UKA on cellular CES1 in living HepG- 2 cells was about 105-fold higher than that in HLMs, which could be attributed to the very poor cell permeability of UKA [41]. These findings suggested that probe 1 could be a reliable and practical tool for label free and high-throughput screening of inhibitors targeting intracellular CES1, which strongly facilitated CES1- associated drug discovery.

|
Download:
|
| Fig. 4. The dose-response inhibition curves of BNPP (a) and UKA (b) towards CES1 activities in recombinant human CES1, HLMs and living cells. | |
{kind=link}
In order to gain a deeper insight into the specificity of probe 1 toward CES1, docking simulations were performed to explore the interactions between probe 1 and CES from the view of ligandenzyme interactions. As shown in Figs. S13 A and B (Supporting information), probe 1 could be well docked into the active site of CES1, while the distances between the ester bond of probe 1 and the key amino acid of catalytic triad (Ser221) and the oxyanion hole (Gly142 and Gly143) of CES1 were within 4 Å. By contrast, although probe 1 could also be docked into the active site of human CES2, the orientation of probe 1 was far away from the oxyanion hole (Gly148 and Gly149) of CES2 (Figs. S13C and D in Supporting information). The distance between the ester bond of probe 1 and Gly149 of CES2 was 7.006 Å (>5.5 Å), implying that probe 1 hydrolysis was hardly hydrolyzed by CES2. These results were consistent with the experimental data and explained why probe 1 could serve as a highly specific probe substrate for CES1.
In summary, a reliable and practical fluorescent probe for highly selective and sensitive sensing CES1 activities in biological systems has been developed and well characterized. Upon addition of CES1- containing samples, probe 1 can be readily hydrolyzed and release corresponding fluorescent metabolite DMBA, which generate robust fluorescence signals around 560 nm and such signals can be easily captured by fluorescence microplate reader and confocal microscopy. The newly developed isoform-specific CES1 probe has been successfully used for real-time monitoring the real activities of CES1 in various biological systems, including tissue preparations, living cells, fresh tissue slices and zebrafish. Furthermore, this probe has also been successfully used for HTS of CES1 inhibitors using living cells as enzyme sources. These findings clearly demonstrate that probe 1 was a practical and broadly applicable tool for highly selective and sensitive sensing CES1 activities in complex biological systems, which holds great promise for further investigations on CES1-associated drug discovery, clinical practice and basic research.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21572029, 31600641, 81703604, 81773687, 81672961 and 81573501), the National Key Research and Development Program of China (Nos. 2017YFC1700200 and 2017YFC1702000), Program of Shanghai Academic/Technology Research Leader (No. 18XD1403600), Shuguang Program (No. 18SG40) supported by Shanghai Education Development Foundation and Shanghai Municipal Education Commission and the Innovative Entrepreneurship Program of High-level Talents in Dalian (Nos. 2016RQ025 and 2017RQ121).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.12.013
| [1] |
S. Bencharit, C.C. Edwards, C.L. Morton, et al., J. Mol. Biol. 363 (2006) 201-214. DOI:10.1016/j.jmb.2006.08.025 |
| [2] |
J.S. Xu, Y.Y. Li, W.D. Chen, et al., Hepatology 59 (2014) 1761-1771. DOI:10.1002/hep.26714 |
| [3] |
D.D. Wang, L.W. Zou, Q. Jin, et al., Acta Pharm. Sin. B 8 (2018) 699-712. DOI:10.1016/j.apsb.2018.05.005 |
| [4] |
E. Dominguez, A. Galmozzi, J.W. Chang, et al., Nat. Chem. Biol. 10 (2014) 113-121. DOI:10.1038/nchembio.1429 |
| [5] |
L.W. Zou, Q. Jin, D.D. Wang, et al., Curr. Med. Chem. 25 (2018) 1627-1649. DOI:10.2174/0929867325666171204155558 |
| [6] |
D.D. Wang, L.W. Zou, Q. Jin, et al., Fitoterapia 117 (2017) 84-95. DOI:10.1016/j.fitote.2017.01.010 |
| [7] |
M.K. Ross, J.A. Crow, J. Biochem. Mol. Toxic. 21 (2007) 187-196. DOI:10.1002/(ISSN)1099-0461 |
| [8] |
T. Satoh, M. Hosokawa, Annu. Rev. Pharmacol. 38 (1998) 257-288. DOI:10.1146/annurev.pharmtox.38.1.257 |
| [9] |
W. Lacour, S. Adjili, J. Blaising, et al., Adv. Healthc. Mater. 5 (2016) 2032-2044. DOI:10.1002/adhm.v5.16 |
| [10] |
L.L. Zhang, H.K. Zhu, C.C. Zhao, et al., Chin. Chem. Lett. 28 (2017) 218-221. DOI:10.1016/j.cclet.2016.07.008 |
| [11] |
G.B. Ge, L. Feng, Q. Jin, et al., Anal. Chim. Acta 989 (2017) 71-79. DOI:10.1016/j.aca.2017.07.048 |
| [12] |
D.F. Yue, M.L. Wang, F. Deng, et al., Chin. Chem. Lett. 29 (2018) 648-656. DOI:10.1016/j.cclet.2018.01.046 |
| [13] |
B.A. Li, Z.S. He, H.X. Zhou, et al., Chin. Chem. Lett. 28 (2017) 1929-1934. DOI:10.1016/j.cclet.2017.08.055 |
| [14] |
Y. Wu, J. Wen, H.J. Li, et al., Chin. Chem. Lett. 28 (2017) 1916-1924. DOI:10.1016/j.cclet.2017.09.032 |
| [15] |
P. Zhang, Z.Q. Guo, C.X. Yan, et al., Chin. Chem. Lett. 28 (2017) 1952-1956. DOI:10.1016/j.cclet.2017.08.038 |
| [16] |
Q. Jin, L. Feng, D.D. Wang, et al., ACS Appl. Mater. Interfaces 7 (2015) 28474-28481. DOI:10.1021/acsami.5b09573 |
| [17] |
L. Feng, Z.M. Liu, J. Hou, et al., Biosens. Bioelectron. 65 (2015) 9-15. DOI:10.1016/j.bios.2014.10.002 |
| [18] |
Q. Jin, L. Feng, D.D. Wang, et al., Biosens. Bioelectron. 83 (2016) 193-199. DOI:10.1016/j.bios.2016.04.075 |
| [19] |
E.A. Halabi, Z. Thiel, N. Trapp, et al., J. Am. Chem. Soc. 139 (2017) 13200-13207. DOI:10.1021/jacs.7b07748 |
| [20] |
L. Liu, C.Q. Sun, J. Yang, et al., Chem.-Eur. J. 24 (2018) 6148-6154. DOI:10.1002/chem.v24.23 |
| [21] |
S.J. Park, Y.J. Kim, J.S. Kang, et al., Anal. Chem. 90 (2018) 9465-9471. DOI:10.1021/acs.analchem.8b02101 |
| [22] |
J.Z. Long, B.F. Cravatt, Chem. Rev. 111 (2011) 6022-6063. DOI:10.1021/cr200075y |
| [23] |
T. Fukami, M. Kariya, T. Kurokawa, et al., Eur. J. Pharm. Sci. 78 (2015) 47-53. DOI:10.1016/j.ejps.2015.07.006 |
| [24] |
Z.M. Liu, L. Feng, G.B. Ge, et al., Biosens. Bioelectron. 57 (2014) 30-35. DOI:10.1016/j.bios.2014.01.049 |
| [25] |
D.D. Wang, Q. Jin, J. Hou, et al., J. Chromatogr. B 1008 (2016) 212-218. DOI:10.1016/j.jchromb.2015.11.046 |
| [26] |
A. Loudet, K. Burgess, Chem. Rev. 107 (2007) 4891-4932. DOI:10.1021/cr078381n |
| [27] |
G. Ulrich, R. Ziessel, A. Harriman, Angew. Chem. Int. Ed. 47 (2008) 1184-1201. DOI:10.1002/(ISSN)1521-3773 |
| [28] |
S. Kim, J. Bouffard, Y. Kim, Chem.-Eur. J. 21 (2015) 17459-17465. DOI:10.1002/chem.201503040 |
| [29] |
M. Taketani, M. Shii, K. Ohura, et al., Life. Sci. 81 (2007) 924-932. DOI:10.1016/j.lfs.2007.07.026 |
| [30] |
L.L. Ding, Z.H. Tian, J. Hou, et al., Acta Pharm. Sin. 52 (2017) 58-65. |
| [31] |
G.B. Ge, J. Ning, L.H. Hu, et al., Chem. Commun. 49 (2013) 9779-9781. DOI:10.1039/c3cc45250f |
| [32] |
Z.R. Dai, L. Feng, Q. Jin, et al., Chem. Sci. 8 (2017) 2795-2803. DOI:10.1039/C6SC03970G |
| [33] |
L.W. Zou, P. Wang, X.K. Qian, et al., Biosens. Bioelectron. 90 (2017) 283-289. DOI:10.1016/j.bios.2016.11.068 |
| [34] |
D.S. Shi, J. Yang, D.F. Yang, et al., J. Pharmacol. Exp. Ther. 319 (2006) 1477-1484. DOI:10.1124/jpet.106.111807 |
| [35] |
Y. Sato, A. Miyashita, T. Iwatsubo, et al., Drug Metab. Dispos. 40 (2012) 1389-1396. DOI:10.1124/dmd.112.045054 |
| [36] |
H.B. Rasmussen, D. Bjerre, K. Linnet, et al., Pharmacogenomics 16 (2015) 649-665. DOI:10.2217/pgs.15.7 |
| [37] |
B.A. Barut, L.I. Zon, Physiol. Genomics 2 (2000) 49-51. DOI:10.1152/physiolgenomics.2000.2.2.49 |
| [38] |
S.P. Choksi, D. Babu, D. Lau, et al., Development 141 (2014) 3410-3419. DOI:10.1242/dev.108209 |
| [39] |
J.M. Nussbaum, L.H.J. Liu, S.A. Hasan, et al., Hepatology 58 (2013) 1326-1338. DOI:10.1002/hep.v58.4 |
| [40] |
N. Fujiyama, M. Miura, S. Kato, et al., Drug Metab. Dispos. 38 (2010) 2210-2217. DOI:10.1124/dmd.110.034249 |
| [41] |
Y. Chen, Q.Q. Wiu, Z.H. Zhang, et al., China J. Chin. Mater. Med. 36 (2011) 988-991. |