 2019, Vol. 30
2019, Vol. 30 


b Beijing Synchrotron Radiation Facility, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China
Recently, a novel route to produce ethanol, which consists of carbonylation of dimethyl ether (DME) to methyl acetate (MA) and hydrogenation of MA to ethanol, has attracted wide attention [1-5]. The major challenge for this novel route is to exploit heterogeneous catalysts for carbonylation of DME with excellently catalytic performance.
Iglesia et al. reported that mordenite (MOR) had the high activity for the carbonylation of DME [6-8]. Many researchers reported that the carbonylation of DME took place at the Brønsted acid (B acid) sites in the 8-MR side pockets of MOR [7, 9]. The high selectivity for the carbonylation of DME in the 8-MR side pockets was due to the proper size of 8-MR side pockets and the unusual orientation of the methoxy group in 8-MR side pockets [9].
Generally, the high catalytic activity tends to cause the deactivation of MOR for carbonylation of DME because of the formation of coke [10]. It becomes a great challenge simultaneously to improve the activity and stability of MOR. The references reported that the acid sites in 12-MR channels of MOR favored to the formation of heavy hydrocarbons, which were responsible for the deactivation [9]. Therefore, reducing the acid sites in 12-MR channels of MOR is an effective way to inhibit the deactivation of MOR. Shen et al. reported that the pre-adsorption of pyridine to cover the acid sites in 12-MR [11] and the removal of the framework Al atoms in 12-MR [12] significantly enhanced the stability of the HMOR catalyst.
As we know, the introduction of metal can significantly affect the acid properties of zeolites. Gabelica et al. prepared MFI zincosilicates by hydrothermal synthesis and proposed that the ZnOH species in Zn-MFI significantly affected the acidity of MFI zeolite [13]. Some researcher reported that the introduction of metal cation by sublimation affected the acidity of zeolites [14]. Li et al. prepared Zn-Co supported ZSM-5 by PEG-additive method, and the acid sites on ZSM-5 changed significantly [15]. Berndt et al. found that in zinc ion-exchanged ZSM-5, Lewis acid (L acid) sites increased, but B acid sites decreased [16].
Herein, we added zinc source to the initial sol during the synthesis of MOR to investigate the effect of zinc on the acid strength, the distribution and the amount of acid sites of MOR. We further investigated the catalytic performance of the Zn-modified MOR for carbonylation of DME.
The X-ray diffraction (XRD) patterns of the HMOR and H-ZnMOR catalysts (Fig. S2 in Supporting information) agree well with the standard MOR (JCPDS No. 11-0155) [17] without any other impurities. The addition of zinc to MOR has little influence on the crystal structure and the intensity of the diffraction peaks of MOR. The high resolution transmission electron microscopy (TEM) images (Fig. S3 in Supporting information) shows that no ZnO particle is on the H-Zn-MOR catalysts. Thus, the XRD and TEM results suggest that the zinc atoms are highly dispersed on the catalysts. We adopted X-ray fluorescence (XRF) to analyze the element composition of the catalysts (Table S1 in Supporting information).
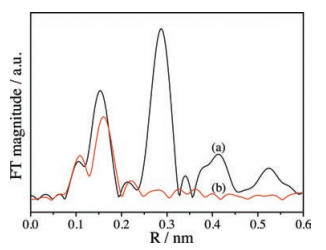
We employed the extended X-Ray absorption fine structure (EXAFS) technique to characterize the information of the local environment of the zinc atoms in the catalysts. Fig. 1 shows the radial distribution function (RDF) profiles derived from the Zn K-edge EXAFS spectra. For the commercial ZnO, the first peak at 0.153 nm corresponds to the Zn-O bond in the ZnO4 tetrahedral arrangement (no correction for the phase displacement scatter) [18]. The second peak at about 0.280 nm belongs to the first Zn-Zn coordination. The peak at 0.420 nm corresponds to the second shell of the Zn-O coordination [19]. For the Zn-0.005 catalyst, the distance of the first Zn-O coordination peak is larger than that for the commercial ZnO. It indicates that the bond length of Zn-O in the Zn-0.005 catalyst is longer than that in the commercial ZnO. We cannot clearly observe the peak corresponding to the first Zn-Zn shell and the second Zn-O shell in the RDF profile of the Zn-0.005 catalyst. It indicates that the zinc atoms in MOR are isolated in the states of single atoms.

|
Download:
|
| Fig. 1. The RDF profiles of (a) ZnO and (b) the Zn-0.005 catalyst. | |
{kind=link}
We carried out the Fourier transform of EXAFS data in the first coordination sphere of zinc and oxygen atoms to get the coordination number and the bond length of Zn-O (Table S2 in Supporting information). The bond lengths of Zn-O in the commercial ZnO and in the Zn-0.005 catalyst are 0.197 nm and 0.207 nm, respectively. Generally, when ZnO4 tetrahedron substitutes for SiO4 into the framework of aluminosilicate (or pure Si) zeolite, the bond length of Zn-O is about 0.196 nm [20]. Therefore, our results show that the Zn atoms do not coordinate into the framework of MOR. According to the EXAFS results, we conclude that the isolated Zn2+ ions exist at the ion-exchange sites in the H-Zn-MOR catalysts.
The Si/Al ratio of zeolite can reflect the amount of acid sites, and the Si/Al ratio of the zeolite framework (defined as Si/Alf) decides the amount of B acid sites. The Si/Al ratio based on the XRF results was defined as Si/Alsum (Table S1). The addition of zinc to MOR leads to a slight increase of Si/Alsum. With the increase of the content of Zn, Si/Alsum gradually increases. Further, we adopted the 27Al magic-angle spinning nuclear magnetic resonance (27Al-MAS NMR) spectra (Fig. S4 in Supporting information) to investigate Si/Alf [21]. The Si/Alf (Table S1) of the H-Zn-MOR catalysts is larger than that of the HMOR catalyst. With the increase of zinc, Si/Alf gradually increases. It indicates that the addition of zinc decreases the coordination of aluminum to the framework of MOR, and further decreases the B acid sites.
The acid properties of the catalysts were detected by temperature-programmed desorption (TPD) of H2O and NH3 with mass spectrometry (MS) as a detector. Fig. 2A shows the MS signal of H2O-TPD. The peaks below 570 K are assigned to the desorption of the physical-adsorption water. The peaks above 570 K are assigned to the dehydration of the B acid sites to form the L acid sites. The peaks of the dehydration of the B acid sites on the HMOR catalyst begins at about 570 K, and two dehydration peaks can be observed. However, for the Zn-0.003 and Zn-0.005 catalysts, only one strong dehydration peak of B acid sites can be observed above 570 K. The dehydration peaks of B acid sites of the Zn-0.01 catalyst are similar to the HMOR catalyst with two dehydration peaks of B acid sites. However, the initial temperature of the dehydration peak of the Zn-0.01 catalyst is at about 660 K, which is higher than that of the HMOR catalyst. The results of H2O-TPD indicate that there are two kinds of B acid sites. The B acid sites, which are relatively easy to dehydrate, are denoted as B-acid-E, and the B acid sites, which are relatively difficult to dehydrate, are denoted as B-acid-D. The addition of Zn to MOR depresses the number of B-acid-E. It is probably because the addition of Zn leads to the change of the Al position in the framework of MOR [22], and further affects the distribution of acid intensity and acid sites in MOR.

|
Download:
|
| Fig. 2. The TPD profiles of (A) H2O and (B) NH3 for the (a) HMOR, (b) Zn-0.003, (c) Zn-0.005 and (d) Zn-0.01 catalysts. | |
{kind=link}
Fig. 2B shows the MS signal of NH3-TPD. The desorption peaks of NH3-TPD are divided into two temperature ranges. The temperature range below 600 K is referred to low-temperature (LT), and the temperature range above 600 K is referred to high-temperature (HT) [23]. Generally, the peaks in the LT range are assign to the desorption of ammonia from weak acid sites [24] and the partial decomposition of NH4+·nNH3 [25]. They are not directly associated with the catalytic activity because the strong acid sites on the 8-member ring of the mordenite are the active sites [9]. One broad asymmetric peak located on the HT range for all the four catalysts. We attribute the peak at about 770 K to the desorption of ammonia from the medium strong acid sites (denoted as Peak 1) and the peak at about 920 K to the desorption of ammonia from the strong acid sites (denoted as Peak 2) [26]. The Peak 1 of the HMOR catalyst is the highest among all the catalysts, indicating that HMOR catalyst has the largest amount of medium strong acid sites among all the catalysts. For the H-Zn-MOR catalysts, with the increase of the content of Zn, the medium strong sites gradually increase. As for the peak 2, the HMOR catalyst has the smallest strong acid sites among all the catalysts. For the H-Zn-MOR catalysts, with the increase of the content of Zn, the strong acid sites gradually decrease. With the addition of zinc into MOR, the total integral area decrease which agrees with the increase of Si/Alsum.
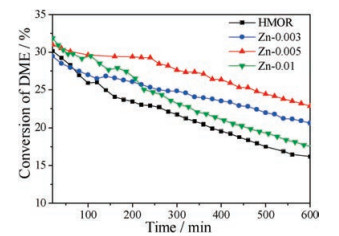
Fig. 3 shows the catalytic performance of carbonylation of DME over the catalysts. For all the catalysts, the conversion of DME reaches the maximum after about 20 min, and then the conversion of DME gradually decreases. The HMOR, Zn-0.005 and Zn-0.01 catalysts exhibit the higher conversion at about 20 min than the Zn-0.003 catalyst. After 10 h, the conversions of DME over the Zn- 0.003 and Zn-0.005 catalysts decrease from 29.2% to 20.6% and from 31.0% to 22.8%, respectively, this is, the conversions decrease about 8.40% for the two catalysts. However, after 10 h, the conversion over the HMOR catalyst decreases from 31.2% to 16.2%, and the conversion of the Zn-0.01 catalyst drops from 31.8% to 17.5%. The drop of the conversion over the two catalysts (about 14.5%) was faster than that over the Zn-0.003 and Zn-0.005 catalysts. The Zn-0.005 catalyst exhibits the best catalytic performance among all the catalysts.

|
Download:
|
| Fig. 3. The activity of carbonylation of DME over the catalysts. The confidence level with error lower than ±3% is 90%. | |
{kind=link}
The references reported that the B acid sites in the 8-MR side pockets of HMOR catalyst were the active sites for carbonylation of DME [27]. The insertion of CO into the middle of the methoxy bonded at the B acid sites was the key step for carbonylation of DME. There is no reference at present showing that the introduction of zinc can form a better active center than B acid sites in the 8-MR side pockets. Generally, the references reported that the activation temperature of CO on the zinc catalysts was above 600 K [28]. We deduce that if Zn2+ ions were in the 8-MR side pockets, they would occupy the space of the 8-MR side pockets to suppress carbonylation of DME due to the small size of the 8-MR side pockets. Therefore, we think that the role of zinc in MOR is not the active sites. According to the 27Al-MAS NMR and TPD results, the introduction of zinc decreases the acid sites in the catalysts. However, compared with the HMOR catalyst, the initial activity of the Zn-0.005 and Zn-0.01 catalyst has little change. We believe that there is no obvious change in the type and quantity of the active centers in the 8-MR side pockets of the catalysts. Thus, we conclude that the introduction of zinc decreases the acid sites in the 12-MR channels, which inhibit the coke deposition and slow down the deactivation of the catalysts. According to the H2O-TPD results, the catalyst with less B-acid-E has higher stability. It indicates that B-acid-E may exist mainly in 12-MR, which can lead to coke deposition.
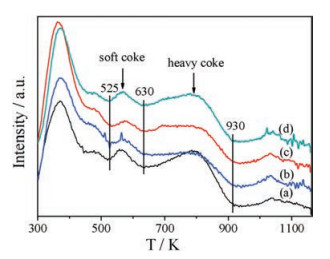
We analyzed the coke deposition in the spent catalysts with the thermo gravimetry (TG) technique. All of the derivative thermo gravimetry (DTG) curves (Fig. 4) are divided into four temperature regions. The weight loss peaks below 525 K correspond to the desorption of physical-adsorb species. The peak between 525 K and 630 K can be attributed to the oxidation of soft coke. The peak between 630 K and 930 K is ascribed to the oxidation of heavy coke [29]. The peak above 930 K might come from the complete dehydroxylation of MOR [30]. Compared with the HMOR catalyst, the H-Zn-MOR catalysts have lower peak intensity of soft coke and heavy coke. Table S3 (Supporting information) lists the weight loss corresponding to the soft and heavy coke of the spent catalysts. The coke deposition of the spent HMOR catalyst is the largest among all the catalysts. After the zinc added into zeolite, the amount of coke deposition reduces. Therefore, the formation of coke during carbonylation of DME is suppressed effectively due to the introduction of zinc to MOR. It suggests that the addition of zinc reduces the active sites for the coke deposition in the 12-MR of MOR.

|
Download:
|
| Fig. 4. The DTG curves of the spent catalysts (a) HMOR, (b) Zn-0.003, (c) Zn-0.005 and (d) Zn-0.01. | |
{kind=link}
In summary, we introduced zinc source into the sols during the synthesis of MOR to prepare the H-Zn-MOR catalysts. The results show that the isolated zinc ions are highly dispersed in MOR. The addition of zinc affects the coordination of aluminum to the framework of MOR. Further, it changes the acid strength and the distribution of acid sites of the catalysts. Carbonylation of DME showed that the initial activity of the Zn-0.005 and Zn-0.01 catalysts was close to that of the HMOR catalyst. After 10 h of carbonylation of DME, the drop of conversion over the HMOR catalyst is nearly two times that over the Zn-0.003 and Zn-0.005 catalysts. The coke deposition on the spent H-Zn-MOR catalysts is less than that on the spent HMOR catalyst. We attribute the enhancement of the catalytic performance of the H-Zn-MOR catalysts to the change of the acid properties.
AcknowledgmentThis work is supported by the National Natural Science Foundation of China (Nos. 21476159, 21676182), the 973 Program (No. 2014CB932403), and authors are also grateful to the Program of Introducing Talents of Disciplines to China Universities (No. B06006).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.05.032.
| [1] |
X.G. San, Y. Zhang, W.J. Shen, N. Tsubaki, Energy Fuels 23 (2009) 2843-2844. DOI:10.1021/ef900080g |
| [2] |
X.G. Li, X.G. San, N. Tsubaki, et al., ChemSusChem 3 (2012) 1192-1199. |
| [3] |
Y. Zhang, X.G. San, N. Tsubaki, Y.S. Tan, J.F. Chen, Ind. Eng. Chem. Res. 49 (2010) 5485-5488. DOI:10.1021/ie901882s |
| [4] |
G.H. Yang, X.G. San, N. Tsubaki, et al., Catal. Today 164 (2011) 425-428. DOI:10.1016/j.cattod.2010.10.027 |
| [5] |
P. Lu, G.H. Yang, Y. Tanaka, N. Tsubaki, Catal. Today 232 (2014) 22-26. DOI:10.1016/j.cattod.2013.10.042 |
| [6] |
A. Bhan, A.D. Allian, G.J. Sunley, D.J. Law, E. Iglesia, J. Am. Chem. Soc. 129 (2007) 4919-4924. DOI:10.1021/ja070094d |
| [7] |
P. Cheung, A. Bhan, G.J. Sunley, E. Iglesia, Angew. Chem. Int. Ed. 45 (2006) 1617-1620. |
| [8] |
P. Cheung, A. Bhan, G.J. Sunley, E. Iglesia, J. Catal. 245 (2007) 110-123. DOI:10.1016/j.jcat.2006.09.020 |
| [9] |
M. Boronat, C. Martı'nez-Sánchez, D. Law, A. Corma, J. Am. Chem. Soc. 130 (2008) 16316-16323. DOI:10.1021/ja805607m |
| [10] |
H.M. Zhan, S.Y. Huang, X.B. Ma, et al., Catal. Sci. Technol. 5 (2015) 4378-4389. DOI:10.1039/C5CY00460H |
| [11] |
J.L. Liu, H.F. Xue, W.J. Shen, et al., Chin. J. Catal. 31 (2010) 729-738. DOI:10.1016/S1872-2067(09)60081-4 |
| [12] |
H.F. Xue, X.M. Huang, E.S. Zhan, M. Ma, W.J. Shen, Catal. Commun. 37 (2013) 75-79. DOI:10.1016/j.catcom.2013.03.033 |
| [13] |
S. Valange, B. Onida, F. Geobaldo, E. Garrone, Z. Gabelica, Stud. Surf. Sci. Catal. 142 (2002) 215-222. DOI:10.1016/S0167-2991(02)80031-3 |
| [14] |
E.I.-M. EI-Malki, R.A. van Santen, W.M.H. Sachtler, J. Phys. Chem. B 103 (1999) 4611-4622. DOI:10.1021/jp990116l |
| [15] |
M. Ammar, Y. Cao, H.Q. Li, et al., Chin. Chem. Lett. 28 (2017) 1583-1589. DOI:10.1016/j.cclet.2017.03.015 |
| [16] |
H. Berndt, G. Lietz, B. Lücke, J. Volter, Appl. Catal. A 146 (1996) 351-363. DOI:10.1016/S0926-860X(96)00092-0 |
| [17] |
H.F. Xue, X.M. Huang, W.J. Shen, et al., Ind. Eng. Chem. Res. 52 (2013) 11510-11515. DOI:10.1021/ie400909u |
| [18] |
A.A. Gabrienko, S.S. Alexander, A.G. Stepanov, et al., ACS Catal. 7 (2017) 1818-1830. DOI:10.1021/acscatal.6b03036 |
| [19] |
L.S. Kau, K.O. Hodgson, E.I. Solomon, J. Am. Chem. Soc. 18 (1989) 7103-7109. |
| [20] |
L. Khouchaf, M.H. Tuilier, M. Wark, M. Soulard, H. Kessler, Microporous Mesoporous Mater. 20 (1998) 27-37. DOI:10.1016/S1387-1811(97)00003-6 |
| [21] |
H. Stach, U. Lohse, B. Parlitz, B. Zibrowius, M. Hunger, J. Phys. Chem. 96 (1992) 8473-8479. DOI:10.1021/j100200a050 |
| [22] |
M. Dong, J.G. Wang, Y.H. Sun, Microporous Mesoporous Mater. 43 (2001) 237-243. DOI:10.1016/S1387-1811(01)00211-6 |
| [23] |
H. Sato, Catal. Rev.-Sci. Eng. 39 (1997) 395-424. DOI:10.1080/01614949708007101 |
| [24] |
Y.H. Liu, N. Zhao, X.G. Li, et al., ACS Appl. Mater. Inter. 7 (2015) 8398-8403. DOI:10.1021/acsami.5b01905 |
| [25] |
F. Lonyi, J. Valyon, Microporous Mesoporous Mater. 47 (2001) 293-301. DOI:10.1016/S1387-1811(01)00389-4 |
| [26] |
M.X. Wang, S.Y. Huang, X.B. Ma, et al., Chin. J. Catal. 37 (2016) 1530-1537. DOI:10.1016/S1872-2067(16)62484-1 |
| [27] |
M. Boronat, C. Martinez, A. Corma, Phys. Chem. Chem. Phys. 13 (2011) 2603-2612. DOI:10.1039/c0cp01996h |
| [28] |
Z.L. Li, J.J. Wang, C. Li, et al., ACS Catal. 7 (2017) 8544-8548. DOI:10.1021/acscatal.7b03251 |
| [29] |
H. Zhou, W.L. Zhu, Z.M. Liu, et al., Catal. Sci. Technol. 5 (2015) 1961-1968. DOI:10.1039/C4CY01580K |
| [30] |
H.G. Karge, V. Dondur, J. Phys. Chem. 94 (1990) 765-772. DOI:10.1021/j100365a047 |