 2019, Vol. 30
2019, Vol. 30 


b Integrated Chinese and Western Medicine Postdoctoral Research Station, Jinan University, Guangzhou 510632, China;
c School of Pharmaceutical Sciences, Guangzhou University of Chinese Medicine, Guangzhou 510006, China;
d Department of Specific Diagnosis, The 324th Military Hospital, Chongqing 400020, China
Wedelia prostrata (Asteraceae), known in China as "Lu Di Ju", is distributed in coastal areas of southern China, such as Taiwan, Fujian, and Guangdong provinces [1]. As a traditional Chinese medicine, the herbs of W. prostrata have been used for the treatment of diphtheria, tonsillitis, pneumonia and bronchitis [1, 2]. According to phytochemical investigations [1, 3, 4], the main chemical constitutes of W. prostrata are terpenoids. Modern pharmacological studies demonstrated that the genus Wedelia possesses various bioactivities, including anti-inflammatory [5-7], anti-bacterial [5, 6, 8], antihepatotoxic [9], anti-viral [10, 11], and antioxidant [5-7, 12] activities.
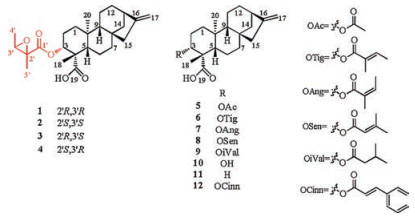
Previously, we studied derivatives of ent-kaurane diterpenes and eudesmanolides from W. prostrata and their antitumor activity [13, 14]. Further investigation of W. prostrata led to the isolation of four new ent-kaurane diterpenes (1-4) and eight known diterpenes (5-12) (Fig. 1). Their structures were elucidated using spectroscopic data, X-ray crystallography, ECD calculations and chemical methods. Moreover, compounds 1-12 were evaluated for their cytotoxicity activities on human HepG-2 cells using the MTT assay. Herein, we report the isolation and structural elucidation of these compounds.

|
Download:
|
| Fig. 1. Chemical structures of diterpenes 1-12. | |
{kind=link}
The air-dried herbs of Wedelia prostrata (7 kg) were powdered and extracted three times with EtOH/H2O (30 L, 95:5, v/v) at room temperature. The concentrated extract (627 g) was suspended in water (4 L) and then extracted with petroleum ether (4 L × 3) and EtOAc (4 L × 3). The PE extract (326 g) was subjected to silica gel column chromatography with a gradient of PE/EtOAc (100:0 to 0:1, v/v) to obtain seven fractions (Fr. 1 - 7). Fr. 3 (17.8 g) was applied to a silica gel CC and eluted with petroleum ether/EtOAc (10:1 to 1:1, v/v) to afford four subfractions (Fr. 3.1 - 3.4). Fr. 3.2 (2.7 g) was purified by Sephadex LH-20 (CHCl3/MeOH, 2:1, v/v) and further separated by preparative HPLC (MeOH/H2O, 80:20, v/v) to yield compound 1 (21.2 mg), 2 (17.5 mg), 3 (19.7 mg) and 4 (16.9 mg). Fr. 3.3 (7.9 g) was subjected to a reversed silica gel CC and eluted with MeOH/H2O (40:60 to 100:0, v/v) to achieve compounds 5 (11.2 mg), 6 (14.1 mg), 7 (15.6 mg) and 8 (12.7 mg). Fr. 3.4 (2.5 g) was separated by preparative HPLC (MeOH/H2O, 70:30, v/v) and further purified by Sephadex LH-20 CC (CHCl3/MeOH, 2:1, v/v) to yield compounds 9 (9.5 mg), 10 (21.3 mg), 11 (21.3 mg) and 12 (13.3 mg).
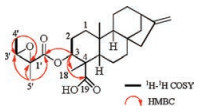
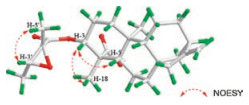
Compound 1 was isolated from CH3OH extract as colorless needles with [α]D25 -45.5 (c 1.0, CHCl3). Its molecular formula was deduced as C25H36O5 on the basis of 13C NMR and HRESIMS data (m/z 439.2476 [M + Na]+, calcd. for C25H36O5: 439.2455). IR spectroscopy suggested the presence of hydroxyl (3458 cm-1) and carbonyl (1734 cm-1) groups. The 1H and 13C NMR data of 1 (Table S1 in Supporting information) indicated the presence of an 2', 3'-epoxyangeloyloxy group [δH 3.04 (CH), 1.55 (CH3), 1.34 (CH3); δC 169.9 (C=O), 60.3 (CH), 60.1 (C), 19.4 (CH3), 13.9 (CH3)] [15], a terminal double bond [δH 4.79 (CH2), 4.73 (CH2); δC 155.5 (C), 103.6 (CH2)] and a carboxyl group (δC 179.4). Besides the above moiety, signals for two methyls, eight methylenes, four methines and three quaternary carbons were observed. The NMR spectroscopic data of 1 resembled those of 3α-tigloyloxykaur-16-en-19-oic acid [16]. The main differences were that the absence of the tigloyloxy group, and the presence of an 2', 3'-epoxyangeloyloxy group in 1 (Fig. 1), which implied that the 3-tigloyloxy group in 3α-tigloyloxykaur-16-en-19- oic acid was replaced by an 2', 3'-epoxyangeloyloxy group in 1. This was confirmed by the 1H-1H COSY cross-peaks between H-3 (δH 4.58) and H2-2 (δH 2.40, 1.72), as well as the HMBC cross-peaks from H-3 to C-1' (δC 169.9)/C-4 (δC 48.9)/C-18 (δC 24.0)/C-19 (δC 179.4) (Fig. 2). The relative stereochemistry of 1 was determined by analysis of the NOESY spectrum. The NOE correlations (Fig. 3) between H-3 (δH 4.58) and H-5 (δH 1.09)/H-18 (δH 1.23) indicated that H-3 was β-orientated [17-19]. Moreover, the NOE correlation between H-3' (δH 3.04) and H3-5' (δH 1.55) indicated that the methyls at C-4' and C-5' were on the opposite side. Furthermore, the absolute configuration of 1 was established by a single-crystal X-ray diffraction experiment. The Cu Kα data resulted in a small Flack parameter of 0.01(7), allowing the assignment of the absolute configuration of 1 as 3R, 4S, 5S, 8S, 9R, 10S, 13R, 2'R, 3'R (Fig. S1 in Supporting information). Based on the above analysis, the structure of 1 was elucidated as shown and named (2'R, 3'R)-3α- (2', 3'-epoxyangeloyloxy)-kaur-16-en-19-oic acid.

|
Download:
|
| Fig. 2. Key 1H-1H COSY, HMBC and NOESY correlations of 1. | |
{kind=link}

|
Download:
|
| Fig. 3. Key NOESY correlations of 1. | |
{kind=link}
Compound 2 was isolated as a white powder with [α]D25 -69.2 (c 1.0, CHCl3). Its molecular formula was deduced as C25H36O5 by HRESIMS data (m/z 439.2472 [M + Na]+, calcd. for C25H36NaO5: 439.2455). The NMR spectroscopic data of 2 was almost identical to those of 1, which indicated that 2 possessed the same planar structure as that of 1. And the relative configuration of 2 was same as that of 1. The β-orientation of H-3 in 2 was determined by the NOE correlations between H-3 (δH 4.63) and H-5 (δH 1.11)/H-18 (δH 1.29) [17-19]. The methyls at C-4' and C-5' of 2 were also located on the opposite side, which was determined by the NOE correlation between H-3' (δH 3.04) and H3-5' (δH 1.54). Interestingly, compound 1 and 2 had the same NMR data, but showed different retention times in HPLC analysis. And the rotation data of 1 (-45.5) and 2 (-69.2) were all negative, indicating they were not a pair of enantiomers. These information implied that compound 1 and 2 could be a pair of diastereoisomers with the oppositely absolute configurations of the 2', 3'-epoxyangeloyloxy groups. The steric configuration of the epoxyangeloyloxy group in 1 was elucidated as (2'R, 3'R) by X-ray diffraction analysis, suggesting the configuration of the epoxyangeloyloxy group of 2 could be supposed to be (2'S, 3'S). In order to confirm the absolute configuration of 2, saponification reactions (Scheme 1) of 1 and 2 were performed. Firstly, compound 1 and 2 were hydrolyzed with NaOH in MeOH/ H2O to give (2R, 3R)- and (2S, 3S)-2, 3-epoxyangelate sodium salt, respectively. Subsequently, neutralization of the sodium salt with HCl in NH4Cl/H2O to yield the corresponding (2R, 3R)- and (2S, 3S)- 2, 3-epoxyangelic acids [15], which were measured the specific rotation values with [α]D25 +28.7 (c 1.0, CHCl3) and -27.2 (c 1.0, CHCl3), respectively. This result was corresponding to the reported values of (2R, 3R)- and (2S, 3S)-2, 3-epoxyangelic acids [15], with [α]D25 +30.0 (c 1.8, CHCl3) and -29.0 (c 1.4, CHCl3), respectively. Therefore, the absolute stereochemistry of the epoxyangelate moiety of 2 was confirmed as (2S, 3S). Moreover, quantumchemical ECD calculations were used to confirm the absolute configuration of 2. The ECD spectra for (3R, 4S, 5S, 8S, 9R, 10S, 13R, 2'S, 3'S)-2 and its enantiomer were performed using the TDDFT-ECD method, and were calculated at the B3LYP/6-31+G (d) level in the gas phase. The experimental ECD spectrum of 2 exhibited a negative cotton effect at 219 nm (△ε -36.7) and a positive cotton effect at 200 nm (△ε +5.6), which were similar to those of the calculated ECD spectrum for (3R, 4S, 5S, 8S, 9R, 10S, 13R, 2'S, 3'S)-2 (Fig. S2 in Supporting information). Hence, the absolute configuration of 2 was elucidated as 3R, 4S, 5S, 8S, 9R, 10S, 13R, 2'S, 3'S. Thus, structure 2 was determined and named (2'S, 3'S)-3α-(2', 3'-epoxyangeloyloxy)-kaur-16-en-19-oic acid.

|
Download:
|
| Scheme 1. Saponification reactions of 1-4. | |
{kind=link}
Compound 3 had the molecular formula C25H36O5 assigned by HRESIMS data (m/z 439.2481 [M + Na]+, calcd. for C25H36NaO5: 439.2455). According to 1D and 2D NMR spectroscopy (Table S2 in Supporting information), compound 3 has the same ent-kaurane diterpene skeleton as 1, except that the chemical shifts of C-1', C-2', C-3', C-4' and C-5' were shifted from δC 169.9, 60.1, 60.3, 13.9, 19.4 in 1 to δC 171.5, 57.8, 58.2, 13.8, 13.4 in 3, respectively. These differences indicated the configuration of 2', 3'-epoxyangeloyloxy group in 3 was different from that of 1. The planar structure of 3 was confirmed by the 1H-1H COSY cross-peaks between H-3 (δH 4.47) and H2-2 (δH 2.33, 1.75), and the HMBC cross-peaks from H-3 to C-1' (δC171.5)/C-4 (δC 48.0)/C-18 (δC 24.0)/C-19 (δC 180.0). The relative configuration of 3 showed similarities to 1 with the β-orientation of H-3, which was indicated by the NOE correlations between H-3 and H-5 (δH 1.09)/H- 18 (δH 1.24) [17-19]. However, based on the NOE correlation between H3-4' (δH 1.33) and H3-5' (δH 1.48), CH3-4' and CH3-5' were determined to be on the same side [20]. To further elucidate the absolute stereochemistry of 3, saponification reactions (Scheme 1) were performed. Comparing the specific rotation data (-6.1) of the hydrolysis product of 3 to reported optical activities of (2R, 3S)- and (2S, 3R)-2, 3-epoxyangelic acids [21], with [α]D25 -5.5 (c 12.0, CHCl3) and +5.5 (c 12.0, CHCl3), the absolute stereochemistry of the epoxyangelate moieties of 3 was confirmed to be (2'R, 3'S). Furthermore, the absolute configuration of 3 was established by quantum-chemical ECD calculations. The predicted ECD spectra of 3, (3R, 4S, 5S, 8S, 9R, 10S, 13R, 2'R, 3'S)-3 (Fig. S3 in Supporting information), and its enantiomer, were compared with the experimental spectra, which exhibited a positive cotton effect at 226 nm (△ε +31.2) and a negative cotton effect at 201 nm (△ε -17.2), indicating the absolute configuration of 3 was 3R, 4S, 5S, 8S, 9R, 10S, 13R, 2'R, 3'S. Accordingly, compound 3 was elucidated and named (2'R, 3'S)-3α-(2', 3'-epoxyangeloyloxy)-kaur-16-en-19-oic acid.
Compound 4 showed an [M + Na]+ ion peak at m/z 439.2470 (calcd. for C25H36NaO5: 439.2455) in the HRESIMS spectrum, consistent with the molecular formula of C25H36O5. The 1D and 2D NMR spectroscopic data of 4 were in strong agreement with those of 3, indicating that the planar structure and relative configuration of compounds 4 were same as those of 3. Similarity to the difference between the absolute configurations of 1 and 2, compounds 3 and 4 could also be a pair of diastereoisomers with opposite absolute configurations of the 2', 3'-epoxyangeloyloxy groups [21]. Saponification (Scheme 1) of 4 gave the corresponding (2S, 3R)-2, 3-epoxyangelic acid, whose specific rotation value was measured to be [α]D25 +6.7 (c 1.0, CDCl3). Comparing the specific rotation data of the hydrolysis product of 4 to reported optical activities of (2R, 3S)- and (2S, 3R)-2, 3-epoxyangelic acids [21], with [α]D25 -5.5 (c 12.0, CHCl3) and +5.5 (c 12.0, CHCl3), the absolute stereochemistry of the epoxyangelate moiety of 4 was confirmed to be (2'S, 3'R). Moreover, the absolute configuration of 4 was determined using quantum-chemical ECD calculations. The experimental ECD spectrum of 4 exhibited a negative cotton effect at 224 nm (△ε -30.1), which was similar to that of the calculated ECD spectrum for (3R, 4S, 5S, 8S, 9R, 10S, 13R, 2'S, 3'R)-4 (Fig. S4 in Supporting information). Hence, the absolute configuration of 4 was elucidated as 3R, 4S, 5S, 8S, 9R, 10S, 13R, 2'S, 3'R. Accordingly, compound 4 was elucidated and named (2'S, 3'R)-3α- (2', 3'-epoxyangeloyloxy)-kaur-16-en-19-oic acid.
The eight known compounds were identified as 3-acetyloxykaurenoic acid (5) [22], 3α-tigloyloxykaur-16-en-19-oic acid (6) [16], 3α-angeloyloxykaur-16-en-19-oic acid (7) [16], 3α-senecionyloxy-ent-kaur-16-en-19-oic acid (8) [23], 3α-isovaleryloxy-ent-kaur-16-en-19-oic acid (9) [23], ent-3β-hydroxy-kaur-16-en-19-oic acid (10) [21], ent-kaur-16-en-19-oic acid (11) [16] and 3α- cinnamoyloxykaur-16-en-19-oic acid (12) [24] (Fig. 1), respectively, by comparing their spectroscopic and physical data with those of related literatures.
The isolates 1-12 were evaluated for their cytotoxic activities on HepG-2 cells using the MTT assay. As shown in Table S3 (Supporting information), all compounds showed cytotoxic activities with IC50 values of 11.72 ± 0.22 μmol/L to 54.75 ± 1.12 μmol/L. Compounds 1-4 showed more potent cytotoxic activities, with IC50 values of 11.72 ± 0.22, 14.23 ± 0.18, 17.35 ± 0.15 and 16.79 ± 0.37 μmol/L, respectively. These results revealed that the presence of epoxyangelate moieties may be related to their cytotoxic activities on HepG-2 cells [25].
In conclusion, four new ent-kaurane diterpenes, which contain (2'R, 3'R)-/(2'S, 3'S)-/(2'S, 3'R)-/(2'R, 3'S)-2', 3'-epoxyangeloyloxy moities, along with eight known ones, were isolated from Wedelia prostrata. Their structures were identified by the spectroscopic data, X-ray crystallography, ECD calculation and chemical methods. Compounds 1-12 were tested for their cytotoxic activities on HepG-2 cells. And all tested isolates show various levels of cytotoxic activities.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (Nos. 81473116, 81673319, 81673670), Science and Technology Planning Project of Guangdong Province (Nos. 2016A030303011, 2016B030301004), Natural Science Foundation of Guangdong Province (No. 2017A030313732), China Postdoctoral Science Foundation (No. 55350202), and Natural Science Foundation of Chongqing (No. cstc2013jcyjA10065).
Appendix A. Supplementary dataSupplementary material related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.05.038
| [1] |
X.G. Zhang, W.Q. Yan, Y.T. Lin, et al., J. Chin. Med. Mat. 34 (2011) 383-386. |
| [2] |
C.L. Li, F.X. Liu, H.Z. Wu, et al., Fujian J. Tradit. Chin. Med. 10 (1959) 448-449. |
| [3] |
S.F. Farag, N.A. El-Emary, M. Niwa, Chem. Pharm. Bull. 44 (1996) 661-664. DOI:10.1248/cpb.44.661 |
| [4] |
C.Y. Ragasa, W.G. Padolina, B.F. Bowden, et al., J. Nat. Prod. 56 (1993) 386-393. DOI:10.1021/np50093a011 |
| [5] |
A. Manjamalai, G.J. Jiflin, G.V.M. Berlin, Asian J. Pharm. Clin. Res. 5 (2012) 155-163. |
| [6] |
M. Govindappa, S.N. Sravya, M.N. Poojashri, et al., J. Med. Plants Res. 5 (2011) 5718-5729. |
| [7] |
R.B.G De, P.R.A. Siqueira, M.L. Kaiser, et al., Lat. Am. J. Pharm. 28 (2009) 594-598. |
| [8] |
A. Manjamalai, V.B.M. Grace, Asian Pac. J. Cancer P. 13 (2012) 3065-3071. DOI:10.7314/APJCP.2012.13.7.3065 |
| [9] |
L.L. Yang, K.Y. Yen, C. Konno, et al., Planta Med. 6 (1986) 499-650. |
| [10] |
F.M. Lin, L.R. Chen, E.H. Lin, et al., Carcinogenesis 28 (2007) 2521-2529. DOI:10.1093/carcin/bgm137 |
| [11] |
P.K. Haldar, S. Bhattacharya, S. Dewanjee, et al., Environ. Toxicol. Phar. 31 (2011) 10-17. DOI:10.1016/j.etap.2010.08.003 |
| [12] |
D. Jayakumar, S.J. Mary, R.J. Santhi, Asian J. Chem. 23 (2011) 305-308. |
| [13] |
Z.N. Wu, Y.B. Zhang, L. Yang, et al., J. Nat. Med. 71 (2017) 305-309. DOI:10.1007/s11418-016-1037-6 |
| [14] |
Z.N. Wu, Y.B. Zhang, N.H. Chen, et al., Chem. Lett. 45 (2016) 1150-1152. DOI:10.1246/cl.160546 |
| [15] |
J.M. Torres-Valencia, C.M. Cerda-Garcia-Rojas, P. Joseph-Nathan, Tetrahedron:Asymmetry 9 (1998) 757-764. DOI:10.1016/S0957-4166(98)00025-1 |
| [16] |
Y. Qiang, D.L. Du, Y.J. Chen, et al., Helv. Chim. Acta 94 (2001) 817-823. |
| [17] |
B.J. Ma, C.N. Wen, Y. Gao, et al., Nat. Prod. Bioprospect. 3 (2013) 107-111. DOI:10.1007/s13659-013-0029-4 |
| [18] |
J.X. Zhang, Y.X. Wang, Z.A. He, et al., Chin. Chem. Lett. 20 (2009) 201-203. DOI:10.1016/j.cclet.2008.10.040 |
| [19] |
G. Ding, J. Wang, J.D. Fei, et al., Chin. Chem. Lett. 28 (2017) 593-596. DOI:10.1016/j.cclet.2016.10.033 |
| [20] |
J.M. Torres-Valencia, C.M. Cerda-Garcia-Rojas, P. Joseph-Nathan, Tetrahedron:Asymmetry 6 (1995) 1611-1616. DOI:10.1016/0957-4166(95)00205-4 |
| [21] |
D.M. Silva, E.V. Costa, P.C.D. Nogueira, et al., Quim. Nova 35 (2012) 1570-1576. DOI:10.1590/S0100-40422012000800015 |
| [22] |
K.Z. Antoine, H. Hussain, E. Dongo, etal., J.AsianNat.Prod.Res. 12 (2010) 629-633. DOI:10.1080/10286020.2010.485933 |
| [23] |
F. Bohlmann, K.H. Knoll, H. Robinsona, et al., Phytochemistry 19 (1980) 107-110. DOI:10.1016/0031-9422(80)85023-0 |
| [24] |
H.S. Vieira, J.A. Takahashi, M.A. Boaventura, Fitoterapia 72 (2001) 854-856. DOI:10.1016/S0367-326X(01)00332-X |
| [25] |
J.M. Torres-Valencia, C.M. Cerda-Garcia-Rojas, L.U. Roman, et al., Phytochemistry 49 (1998) 2569-2572. DOI:10.1016/S0031-9422(98)00304-5 |