 2019, Vol. 30
2019, Vol. 30 


Panax notoginseng (Burk.) F.H. Chen is a species of the genus Panax in the family Araliaceae and is widely cultivated in Yunnan and Guangxi provinces of China. As the key components of this plant [1], more than 150 saponins were obtained from its roots, leaves, flower buds and seeds [2-4]. Pharmacological experiments showed that P. notoginseng saponins have various interesting bioactivities, such as anti-diabetes [5], repairing the function of impaired nerves [6], preventing atherosclerosis [7], and alleviating osteoporosis [8]. Previously, we also reported eleven dammaranetype saponins called notoginsenosides Ng1-Ng2 and Fh1-Fh9 from the leaves of P. notoginseng [9-11]. A continuation of research on the same herb led to the isolation of two new dammarane-type saponins, named notoginsenoside-Ng3 (1) and notoginsenoside-Ng4 (2), together with three known saponins, notoginsenoside Fa (3) [12], notoginsenoside LK2 (4) [13] and notoginsenoside LK4 (5) [13] (Fig. 1). Herein, we present the isolation and structure determination of five saponins (1–5) from this plant as well as the inhibitory activities against the acetylcholinesterase (AchE).

|
Download:
|
| Fig. 1. Compounds 1–5 isolated from the leaves of Panax notoginseng. | |
The 25 kg air-dried leaves of P. notoginseng were successively extracted with 95% EtOH (150 L × 2) and water (150 L × 2). The water extract was loaded on a D101 macroporous absorbent resin column (100 cm × 20 cm) and eluted with EtOH/H2O (0, 25%, 50%, and 95%). The 50% EtOH fraction (365.4 g) was further subjected to a diatomite column [EtOAc, acetone, acetone/EtOH (1:1, v/v), EtOH/ H2O (7:3, v/v), EtOH/H2O (1:1, v/v)] to obtain 5 fractions. The acetone/EtOH (1:1, v/v) fraction (146 g) was applied to a silica gel column, MPLC system and preparative HPLC to give compounds 1 (29 mg), 2 (6 mg), 3 (55 mg), 4 (8 mg), and 5 (5 mg).
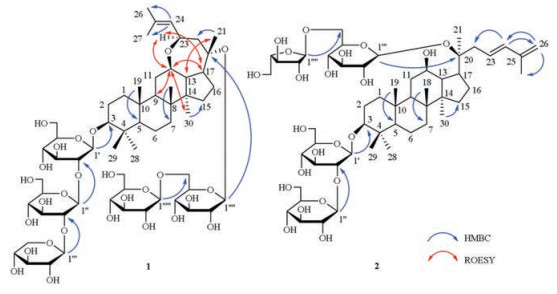
Notoginsenoside-Ng3 (1)was obtained as white amorphous powder. [α]D20 -6.0 (c 0.1, MeOH); UV λmax (MeOH, nm) (logε): 203 (2.79), 255 (2.29); IR (KBr, cm-1) νmax: 3429 (OH), 2920 (CH), 1628 (C=C), 1076 (C-O-C). Compound 1 showed an ion peak at m/z 1261.6147 (calcd. for [M+Na]+: 1261.6188) on the basis of HRESIMS, and its molecular formula was confirmed as C59H98O27. The 1H NMR data (see Table 1) of 1 presented eight methyl protons at δH 0.80, 0.91, 1.04, 1.10, 1.26, 1.51, 1.67 and 1.81, and an olefinic proton signal at δH 5.54 (d, 1H, J = 7.8 Hz). In the 13C NMR data (see Table 1) of 1, two olefinic carbons were observed at δC 129.2 and 131.2; five anomeric carbons were detected at δC 99.2, 103.1, 104.7, 105.5 and 106.4. In view of above mentioned characteristic peaks, compound 1 was superimposable on the aglycone of epoxynotoginsenoside A [14], while the carbon signals of the sugar moieties were similar to notoginsenoside Fh4 [11]. In the ROESY spectrum (Fig. 2), correlations between H-12 (3.63, m, 1H) and H-9 (1.46, overlapped, 1H)/H-17 (3.12, td, 1H, J = 10.2, 4.2 Hz)/Me-30 (1.04, s, 3H), as well as correlations between H-23 and H-12/H-17 indicated the α-orientation of H-12 and H-23; the correlations between H-21 (1.51, s, 3H) and H-13 (1.57, t, 1H, J = 10.2 Hz), as well as between H- 13 and H-18 (0.91, s, 3H), presented the configuration of C-21 was β. The acid hydrolysis of compound 1 presented glucose and xylose through TLC by comparing with authentic sugars. According to GC analysis of their trimethylsilyl ether derivatives (Supporting information), the sugar moieties were determined as D-glucose (tR: 20.45 min) and D-xylose (tR: 14.85 min). Their relative configuration were defined as β by the coupling constants (J) at H-1' (4.93, d, 1H, J = 7.8 Hz), H-1" (5.51, d, 1H, J = 7.8 Hz), H-1"' (5.40, d, 1H, J = 7.2 Hz), H-1"" (5.09, d, 1H, J = 7.8 Hz) and H-1"" (5.10, d, 1H, J = 7.8 Hz). Furthermore, in HMBC spectrum (Fig. 2), the correlations between H-1' and C-3 (88.8), H-1" and C-2' (82.9), H-1"' and C-2" (84.5), H-1"" and C-20 (82.0), H-1"" and C-6"" (70.4)suggested the positions and sequences of the sugar moieties; an ether linkage between C-12 and C-23 was observed by the long correlations between H-23 (4.82, t, 1H, J = 8.4 Hz) and C-12 (79.7). With the help of DEPT, 1H-1H COSY, HSQC, HMBC, ROESYand TOCSY spectra (Supporting information), all the 13C and 1H NMR data of compound 1 were assigned and listed in Table 1.
|
|
Table 1 13C NMR (150 MHz) and 1H NMR (600 MHz) data of compounds 1–3 in pyridine-d5. |

{kind=link}

|
Download:
|
| Fig. 2. The key HMBC and ROESY correlations of compounds 1 and 2. | |
{kind=link}
Therefore, the structure of compound 1 was concluded to be 3β, 20S-dihydroxy-12β, 23R-epoxydammar-24-ene 3-O-β-D-xylopyranosyl-(1 → 2)-β-D-glucopyranosyl-(1 → 2)-β-D-glucopyranosyl-20-O-β-D-glucopyranosyl-(1 →6)-β-D-glucopyranoside (Fig. 1).
Notoginsenoside-Ng4 (2) was obtained as white amorphous powder. [α]D20 +9.0 (c 0.1, MeOH); UV λmax (MeOH, nm) (logε): 229 (2.98); IR (KBr, cm-1) νmax: 3427 (OH), 2970 (CH), 1632 (C=C), 1455 (conjugated C=C). Compound 2 showed an ion peak at m/z 1099.5680 (calcd. for [M+Na]+: 1099.5659) on the basis of HRESIMS, and its molecular formula was confirmed as C53H88O22. When 1H NMR data (Table 1) of 2 showed seven methyl protons at δH 0.84, 0.88, 1.02, 1.12, 1.30, 1.61 and 1.95, their carbon signals were also observed at δC 16.3, 17.1, 15.9, 16.6, 28.1, 23.5 and 19.0 in 13C NMR data (see Table 1), respectively. With the aid of HSQC spectrum (Fig. S23 in Supporting information), two pair of olefinic carbons were observed at δC 114.9, 127.4, 135.7 and 142.6, and their positions were deduced by the H-C correlations between two germinal proton signals at H-26 (4.95, overlapped, 1H)/(5.03, overlapped, 1H) and C-24 (135.7)/C-27 (19.0), as well as H-24 (6.49, d, 1H, J = 15.0 Hz) and C-22 (40.2)/C-26 (114.9) in HMBC spectrum (Fig. 2). Comparing the 13C NMR data for 2 with reference values, the carbon signals matched with the aglycone of quinquenoside L1 [15], while the sugar chains were similar to those of ginsenoside Rc [16]. In addition, L-arabinose and D-glucose of compound 2 were confirmed by the acid hydrolysis and GC analysis (tR: L-arabinose, 14.86 min; D-glucose, 20.42 min). And the correlations in HMBC spectrum (Fig. 2) between H-1' (4.95, overlapped, 1H) and C-3 (89.0), H-1" (5.39, d, 1H, J = 7.8 Hz) and C-2'(83.5), H-1"' (5.18, d, 1H, J = 7.8 Hz) and C-20 (83.3), H-1"' (5.68, s, 1H) and C-6"' (68.3) revealed the positions and sequences of the sugar moieties.
From the foregoing evidences, compound 2 was confirmed as 3β, 12β, 20S-trihydroxydammar-23, 25-diene 3-O-β-D-glucopyranosyl-(1 → 2)-β-D-glucopyranosyl-20-O-α-L-arabinofuransyl- (1 →6)-β-D-glucopyranoside (Fig. 1).
Notoginsenoside Fa (3) was obtained as a block crystal (in MeOH), and gave an ion peak at m/z 1263.6313 (calcd. for [M+Na]+: 1263.6344) in HRESIMS, indicating the formula of 3 to be C59H100O27. In 1H NMR data (Table 1), eight methyl protons were observed at δH 0.80 (CH3), 0.96 (CH3), 0.97 (CH3), 1.11 (CH3), 1.28 (CH3), 1.61 (CH3) and 1.66 (2CH3); five anomeric protons were presented at δH 4.93 (d, 1H, J = 7.8 Hz), 5.11 (d, 1H, J = 7.8 Hz), 5.13 (d, 1H, J = 7.8 Hz), 5.41 (d, 1H, J = 7.2 Hz) and 5.53 (d, 1H, J = 7.8 Hz). 13C NMR data (Table 1) showed a pair of olefinic carbons at δC 126.0 and 131.1, as well as five anomeric carbons at δC 98.1, 103.2, 104.8, 105.4 and 106.5. According to the comprehensive analysis of 1D and 2D NMR spectra, compound 3 was deduced to be notoginsenoside Fa [12]. And a single crystal of 3 was obtained, thus the absolute configuration of 3 was unquestionably elucidated as described in Fig. 1. The single crystal structure of notoginsenoside Fa was showed in Fig. 3. X-ray crystallographic data for 3: C59H100O27·3MeOH, M = 1337.52, orthorhombic system, space group P212121 (No. 19), Z = 4, μ(Cu Kα) = 0.882 mm-1, Flack parameter -0.07 (8). The data can be accessed via www.ccdc.cam.ac.uk (CCDC: 1817469).

|
Download:
|
| Fig. 3. X-ray crystal structure of compound 3. | |
{kind=link}
By compared the NMR and MS spectra of compounds 4 and 5 with literature data of the corresponding compounds [13], another two saponins were identified as notoginsenoside LK2 (4) and notoginsenoside LK4 (5).
AchE inhibition experiments were also conducted on the method in our previous reported [17], all the tested compounds (1–5) showed weak inhibitory activity against the AchE, with inhibitory ratio of 28.8%, 30.4%, 16.2%, 27.5% and 21.6% at the final concentration of 0.16 mmol/L, respectively. The positive control (Galanthamine) showed 91.9% inhibitory profile at a dose of 0.04 mmol/L.
The phytochemical study of P. notoginseng leaves led to the isolation of five saponins including two new ones, notoginsenoside-Ng3 (1)and notoginsenoside-Ng4 (2), along with three known saponins (3–5). Bioactive experiment indicated that compounds 1–5 showed weak inhibitoty activities against AchE. These results enrich the chemical and bioactive investigations of P. notoginseng leaves. The single crystal data of notoginsenoside Fa was reported for the first time, which provided scientific values for the study of saponins.
AcknowledgmentsThis research program was financially supported by the National Natural Science Foundation of China (Nos. 81630094 and 81730093) and CAMS Innovation Fund for Medical Sciences (CIFMS) (No. 2016-I2M-2-003). The author would like to thank Fangyou Chen and Ying Chen from IMM, CAMS&PUMC, for reviewing this paper. We are truly grateful to the Department of Instrumental Analysis, IMM, CAMS&PUMC for measuring MS, UV and NMR spectra.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2018.04.015.
| [1] |
W. Xu, C.F. Ding, X.W. Wang, et al., Chin. Chem. Lett. 26 (2015) 1298-1302. DOI:10.1016/j.cclet.2015.07.014 |
| [2] |
X.Y. Wang, D. Wang, X.X. Ma, et al., Helv. Chim. Acta 91 (2008) 60-66. |
| [3] |
T. Wang, R.X. Guo, G.H. Zhou, et al., J. Ethnopharmacol. 188 (2016) 234-258. DOI:10.1016/j.jep.2016.05.005 |
| [4] |
C.Z. Gu, Y.J. Qiao, D. Wang, et al., Nat. Prod. Res. 32 (2017) 294-301. |
| [5] |
R. Uzayisenga, P.A. Ayeka, Y. Wang, Phytother. Res. 28 (2014) 510-516. DOI:10.1002/ptr.v28.4 |
| [6] |
L.X. Liu, L.Q. Zhu, Y.H. Zou, et al., Chem. Pharm. Bull. 37 (2014) 560-568. DOI:10.1248/bpb.b13-00770 |
| [7] |
J. Li, Z.Z. Xie, Y.B. Tang, et al., Eur. J. Pharmacol. 652 (2011) 104-110. DOI:10.1016/j.ejphar.2010.11.017 |
| [8] |
Y. Shen, Y.Q. Li, S.P. Li, et al., J. Nat. Med. 64 (2010) 336-345. DOI:10.1007/s11418-010-0416-7 |
| [9] |
J.W. Huang, F.Y. Chen, C.J. Li, et al., J. Asian Nat. Prod. Res. 20 (2018) 337-343. DOI:10.1080/10286020.2017.1403421 |
| [10] |
X.Y. Liu, J. Ma, C.J. Li, et al., Acta Pharm. Sin. 52 (2017) 1432-1436. |
| [11] |
X.Y. Liu, S. Wang, C.J. Li, et al., Phytochemistry 145 (2017) 10-17. |
| [12] |
T.R. Yang, R. Kasai, J. Zhou, Phytochemistry 22 (1983) 1473-1478. DOI:10.1016/S0031-9422(00)84039-X |
| [13] |
J.Li, R.F.Wang, Y.Zhou, et al., J.Ginseng Res.(2017), doi: [DOI:http://dx.doi.org/10.1016/j.jgr.2017.11.008].
|
| [14] |
C. Yuan, F.X. Xu, X.J. Huang, et al., Chin. J. Nat. Med. 13 (2015) 303-306. |
| [15] |
J.H. Wang, W. Li, Y. Sha, et al., J. Asian Nat. Prod. Res. 3 (2001) 123-130. DOI:10.1080/10286020108041379 |
| [16] |
J.G. Cho, M.K. Lee, J.W. Lee, et al., J. Ginseng Res. 34 (2010) 113-121. DOI:10.5142/jgr.2010.34.2.113 |
| [17] |
Y.B. Tu, J.W. Huang, Y.F. Li, Med. Chem. Res. 27 (2018) 857-863. DOI:10.1007/s00044-017-2108-2 |