 2019, Vol. 30
2019, Vol. 30 


b College of Pharmacy, Guangdong Pharmaceutical University, Guangzhou 510006, China;
c Program for Natural Products Chemical Biology, Key Laboratory of Plant Resources Conservation and Sustainable Utilization, Guangdong Provincial Key Laboratory of Applied Botany, South China Botanical Garden, Chinese Academy of Sciences, Guangzhou 510650, China
Marine natural products have been identified as one of the most ubiquitous and significant treasure-trove in the repertoire of drug discovery, the broadly bioactive nature of which is further reflected by its accessibility of impressive structural diversity and promising pharmacological effect in medicinal industry [1, 2]. It is also noteworthy that more than 30 marine-derived natural products hold a promising potential in the treatment of various cancers and have been widely applied in clinical or preclinical trails [3, 4]. Advancement of a bioactive marine natural product with therapeutic potential is greatly facilitated by considerable endeavors from the synthetic and pharmaceutical communities on the discovery of novel compounds from marine fungi [5-7]. Marinederived Phomopsis fungi, are well-known as one of the most productive fungal taxa with the ability to generate structurally novel and architecturally diverse secondary metabolites, many of which displayed a broad spectrum of fascinating biological activities including BACE1 inhibitory [8], immunosuppressive [9], antifungal [10], anti-inflammatory [11], antitumor [12], antimicrobial [13], and antimigratory activities [14].
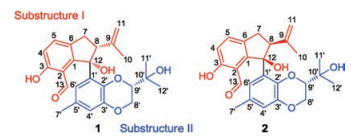
Stimulated by the fruitful achievements on the natural product chemistry from marine fungi, our group has been dedicated to the discovery of novel and structural diverse antitumor compounds from marine-derived fungi [15-19]. Recently, the EtOAc extract of the solid-substrate fermentation of the fungus Phomopsis lithocarpus FS508, which was isolated from a sediment sample collected from Indian Ocean, was analyzed. Bioassay-guided separation was customized for the isolation of a pair of novel tenellone diastereoisomers, named as lithocarpinols A (1) and B (2), both of which shared an unprecedented 2, 3-dihydro-1Hindene fused architecture and represented a new class of tenellone family (Fig. 1) [20]. Herein, the details of isolation, structural elucidation by NMR spectral interpretation, X-ray diffraction and quantum molecular calculation and biological evaluation were described.

|
Download:
|
| Fig. 1. The structures of compounds 1 and 2. | |
Lithocarpinol A (1) was isolated as a white powder and its molecular formula was assigned as C25H28O6 based on the negative high-resolution ESI mass spectrum, wherein a deprotonated molecular ion was observed at m/z 423.1815 ([M–H]-, calcd. for C25H27O6: 423.1813), implying the presence of 12 indices of hydrogen deficiency. The IR spectrum (Fig. S10 in Supporting information) of 1 showed characteristic absorption bands for hydroxy (3373 cm-1) and carbonyl (1643 cm-1) functionalities, respectively. Moreover, the 13C NMR data (Table 1) in conjunction with the HSQC spectrum (Fig. S4 in Supporting information) of 1 further successfully confirmed the existence of 25 carbon resonances corresponding to four methyl groups, two methylenes, seven methines, and twelve quaternary carbons including one carbonyl one. Moreover, the 1H-1H COSY and HSQC spectra unambiguously disclosed the presence of three spin coupling systems as depicted with bold blue lines in Fig. 2: a (H-4/H-5), b (H-7/H-8), and c (H-8'/H-9').
|
|
Table 1 1H (500 MHz) and 13C (125 MHz) NMR data of 1 in CD3COCD3. |


|
Download:
|
| Fig. 2. 1H-1H COSYs and key HMBCs of 1 and 2. | |
The establishment of the planar structure for 1 was initially conducted by the interpretation of the 2D NMR, in particular the HMBC correlations (Fig. S6 in Supporting information). Firstly, the HMBC correlations from H-4 to C-2 and C-6, as well as H-5 to C-1 and C-3 successfully accompanied the establishment of the 1, 2, 3, 6- tetrasubstituted aromatic ring based on the aforementioned deductive fragment a. The linkage between aldehyde substitution C-13 and aromatic ring A was strongly suggested to be located at C- 2, which could be rationally verified by the HMBC correlations of the aldehyde proton (δH 9.73) to C-2 and C-3. Moreover, the phenolic proton (3-OH, δH 11.55) could be rationally assigned as ortho to aldehyde moiety, giving rise to a strong intramolecular hydrogen bonding effect and causing the signal of 3-OH to be significantly downfield, and it was finally anchored at C-3 position evidenced by the critical HMBC correlations from 3-OH (δH 11.55) to C-3 and C-4. As referring to fragment b, the ring B was assumed to be fused with aromatic ring A through C-1 and C-6 to form a novel cyclopentane ring system, this conclusive deduction could be further rationalized by the aid of the HMBC correlations from H-7 to C-1, C-6 and C-12 as well as H-8 to C-1 and C-12. Furthermore, HMBC interactions of H-10 and H-11 to C-8 and C-9 adequately attributed the location of 2-propene at C-8 in the cyclopentane ring B. Therefore, the substructure I was completely determined and depicted as shown in Fig. 2.
In the substructure Ⅱ, the meta-coupled aromatic protons of H- 4' and H-6' implied a 1', 2', 3', 5'-tetrasubstituted benzene ring C, which could be further confirmed and supported by the obvious HMBC interactions from H-4' to C-2', C-3' and C-6'. The linkage of methyl group and the ring C could be reasonably assigned at C-5' (δC 130.7) position attributable to the HMBC correlations from H-7' to C-4', C-5' and C-6'. Moreover, the location of the two oxygen atoms at C-2' and C-3' mainly referred to their significant downshifted carbon signals at δC 138.6 and 144.6. The HMBC cross peak of H-80 (δH 3.90, 4.42) to C-3' (δC 144.6) strongly indicated that the benzene ring ought to be fused with the fragment c via C-2' and C- 3' to constitute a 2, 3-dihydrobenzo-[b][1,4]dioxane architecture. The position of propan-2-ol moiety could be readily assigned at C- 90 position because of the pivotal HMBC interactions from H-11' and H-12' to C-9' and C-10'. Consequently, the substructure Ⅱ was ascertained. With a careful inspection and interpretation of HMBC spectrum, the conclusive correlations from H-8 (δH 3.77) to C-10 (δC 135.6) and C-12 (δC 84.5) were successfully distinguished, which unambiguously verified the direct linkage of substructures Ⅰ and Ⅱ through C-12 accompanied with formation of a tertiary hydroxyl functionality. Therefore, compound 1 was elucidated as a novel tenellone derivative featuring an unprecedented 2, 3-dihydro-1H-indene fused-ring system.
compound 2 was obtained as a white powder. The molecular formula was assigned as C25H28O6 based on the positive mode HRESI-MS m/z 447.1777 ([M + Na]+, calcd. for C25H28O6Na: 447.1778), indicating the presence of twelve degrees of unsaturation. A closer comparison of its 1H and 13C NMR spectra with those of 1 suggested that they should share the same planar structure due to the similarity of the 1H and 13C NMR profiles in most cases (Table 2), which was re-confirmed by the same HMBC correlations as depicted in Fig. 2. However, subtle differences were detected with the chemical shifts between 1 and 2, particularly the chemical shifts at H-8, H-13, H-6', H-8'b, H-9', H-11' (δH 3.77, 9.73, 7.14, 3.91, 3.54 and 0.90 for 1, and δH 3.59, 9.82, 7.07, 3.70, 3.79 and 0.99 for 2), indicating that 1 and 2 should be a pair of diastereoisomers.
|
|
Table 2 1H (500 MHz) and 13C (125 MHz) NMR data of 2 in CD3COCD3. |
Topologically, the presence of a key stereogenic center (C-9') far away from the 2, 3-dihydro-1H-indene core attenuated the longrange correlations, thus the prospect of assigning their relative configurations by NMR spectra for compounds 1 and 2 seemed rather bleak. In this regard, effort towards conclusive evidence referring to single-crystal X-ray diffraction experiment was conducted with extensive screening of solvent combinations, whereas all the attempts to obtain a single crystal from pure compound 1 or 2 proved to be invalid. Amazingly, when a mixture (1:1) of compounds 1 and 2 was crystallized together from methanol solution, a cocrystal was obtained, which beautifully set the stage for an efficacious X-ray diffraction analysis and successfully removed the bottleneck for assigning their stereogenic centers.
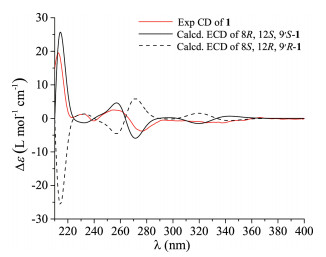
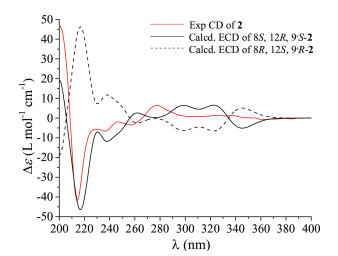
Thus, the X-ray crystallographic experiment was performed using Cu Kα with a Flack parameter of 0.08(6), and the absolute configurations of the two compounds were then unambiguously determined as 8R, 12S, 9'S and 8S, 12R, 9'S (Fig. 3). However, how to differentiate the compounds 1 and 2 from the cocrystal became another problem. To address this issue, the electronic circular dichroism (ECD) calculation using the time-dependent DFT was performed. As expected, the calculated ECD spectrum for 8R, 12S, 9'S was perfectly matched to the experimental one of compound 1 (Fig. 4), while the theoretical CD curve of the 8S, 12R, 9'S isomer agreed well with the experimental one of compound 2 (Fig. 5). Consequently, the absolute configurations of compounds 1 and 2 were finally assigned to be 8R, 12S, 9'S and 8S, 12R, 9'S, respectively.

|
Download:
|
| Fig. 3. Perspective drawing of the X-ray structures of compounds 1 (left) and 2 (right). | |

|
Download:
|
| Fig. 4. Experimental and calculated ECD spectra of 1. | |

|
Download:
|
| Fig. 5. Experimental and calculated ECD spectra of 2. | |
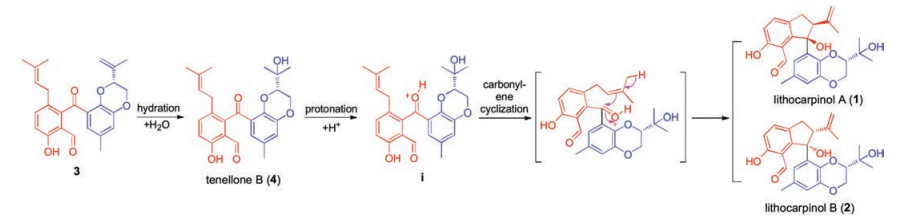
To the best of our knowledge, lithocarpinols A and B represented the first class of natural tenellone with an unprecedented 2, 3-dihydro-1H-indene core, and they existed as a pair of diastereoisomers [20]. With an elaborate and deep insight into the innate chemical properties of their skeleton, Scheme 1 outlines, in biosynthetic format, a plausible biogenetic pathway hinged on a fascinating key carbonyl-ene cyclization to account for their biosynthetic origination. Briefly, they might biogenetically derive from the known tenellone 4 [21], which was also isolated with more than 300 mg in this fungus. Initial hydration of the olefin moiety in 3 would generate the proposed biogenetic precursor 4. The further protonation of 4 afforded the key oxonium intermediate (i), providing advantageous conditions for a vital spontaneous carbonyl-ene cyclization implemented by the pendant isopentene moiety, thus leading to the construction of the 2, 3-dihydro-1H-indene architecture and generation of a pair of diastereoisomers, lithocarpinols A and B.

|
Download:
|
| Scheme 1. Proposed biogenetic pathway of lithocarpinols A and B. | |
Lithocarpinols A and B were evaluated for their cytotoxicities against four human tumor cell lines HepG-2, MCF-7, SF-268, and NCI-H460 with cisplatin as the positive control (Table 3). Both of them showed inhibitory activity against all the tested cell lines; especially, lithocarpinol A (1) exhibited moderate inhibitory effect against HepG-2 and A549 cell lines with IC50 values of 9.4 μmol/L and 10.9 μmol/L, respectively. However, compound 2 just displayed weak inhibitory activity with IC50 values in the range of μmol/L.
|
|
Table 3 Cytotoxic activities of compounds 1 and 2. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In conclusion, a pair of novel tenellone diastereoisomers, lithocarpinols A and B, were isolated and structurally elucidated from the deep-sea-derived fungus P. lithocarpus FS508. They are the first reported examples of tenellone derivatives possessing an unprecedented 2, 3-dihydro-1H-indene core. Their biosynthetic speculation featured a very enticing carbonyl-ene cyclization, which might illuminate an actionable roadmap to the biosynthetic pathway proposal of indene derivatives. Moreover, the cytotoxic experiment indicated that the bioactive compound lithocarpin A warrants further total synthesis and biological evaluation endeavors.
AcknowledgmentsThis work was financially supported by the Science and Technology Program of Guangzhou, China (No. 201607020018), the Team Project of the Natural Science Foundation of Guangdong Province (No. 2016A030312014), the National Natural Science Foundation of China (No. 31272087), the Guangdong Provincial Project for Science and Technology (Nos. 2015A030302061, 2016A020222022), and the Guangdong Provincial Innovative Development of Marine Economy Regional Demonstration Projects (No. GD2012-D01-002). We sincerely thank Mr. Can Li of Central Laboratory of Southern Medical University for NMR measurement.
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.09.018.
| [1] |
P.E. Jans, A.M. Mfuh, H.D. Arman, et al., J. Nat. Prod. 80 (2017) 676-683. DOI:10.1021/acs.jnatprod.6b00963 |
| [2] |
J.W. Blunt, A.R. Carroll, B.R. Copp, et al., Nat. Prod. Rep. 35 (2018) 8-53. DOI:10.1039/C7NP00052A |
| [3] |
Z.Q. Xiong, J.F. Wang, Y.Y. Hao, et al., Mar. Drugs 11 (2013) 700-717. DOI:10.3390/md11030700 |
| [4] |
D.J. Newman, G.M. Cragg, J. Nat. Prod. 79 (2016) 629-661. DOI:10.1021/acs.jnatprod.5b01055 |
| [5] |
X.D. Li, X.M. Li, G.M. Xu, et al., J. Nat. Prod. 78 (2015) 844-849. DOI:10.1021/acs.jnatprod.5b00004 |
| [6] |
C. Wang, L. Guo, J.J. Hao, et al., J. Nat. Prod. 79 (2016) 2977-2981. DOI:10.1021/acs.jnatprod.6b00766 |
| [7] |
L.J. Liao, S.Y. Bae, T.H. Won, et al., Org. Lett. 19 (2017) 2066-2069. DOI:10.1021/acs.orglett.7b00661 |
| [8] |
S.S. Xie, Y. Wu, Y.B. Qiao, et al., J. Nat. Prod. 81 (2018) 1311-1320. DOI:10.1021/acs.jnatprod.7b00889 |
| [9] |
W. Wei, J. Gao, Y. Shen, et al., Eur. J. Org. Chem. 2014 (2014) 5728-5734. DOI:10.1002/ejoc.201402491 |
| [10] |
H. Hussain, K. Krohn, I. Ahmed, et al., Eur. J. Org. Chem. 2012 (2012) 1783-1789. |
| [11] |
Z.X. Hu, Y. Wu, S.S. Xie, et al., Org. Lett. 19 (2017) 258-261. DOI:10.1021/acs.orglett.6b03557 |
| [12] |
Z. Shang, R. Raju, A.A. Salim, et al., J. Org. Chem. 82 (2017) 9704-9709. DOI:10.1021/acs.joc.7b01793 |
| [13] |
M.X. Huang, J. Li, L. Liu, et al., , Mar. Drugs 14 (2016) 215. DOI:10.3390/md14110215 |
| [14] |
B.C. Yan, W.G. Wang, D.B. Hu, et al., Org. Lett. 18 (2016) 1108-1111. DOI:10.1021/acs.orglett.6b00214 |
| [15] |
J.L. Xu, H.B. Tan, Y.C. Chen, et al., Org. Chem. Front. 5 (2018) 1792-1797. DOI:10.1039/C8QO00095F |
| [16] |
Z. Fan, Z.H. Sun, Z. Liu, et al., Mar. Drugs 14 (2016) 164. DOI:10.3390/md14090164 |
| [17] |
X.W. Gao, H.X. Liu, Z.H. Sun, et al., Molecules 21 (2016) 371. DOI:10.3390/molecules21040371 |
| [18] |
H.X. Liu, L. Zhang, Y.C. Chen, et al., Nat. Prod. Res. 31 (2017) 404-410. DOI:10.1080/14786419.2016.1169418 |
| [19] |
H.X. Liu, L. Zhang, Y.C. Chen, et al., J. Asian Nat. Prod. Res. 19 (2017) 145-151. DOI:10.1080/10286020.2016.1189906 |
| [20] |
Z.G. She, H. Cui, Y.H. Pan, et al., Patent, CN 108277164 A, 2018.
|
| [21] |
C.W. Zhang, J.G. Ondeyka, K.B. Herath, et al., J. Nat. Prod. 68 (2005) 611-613. DOI:10.1021/np049591n |