 2019, Vol. 30
2019, Vol. 30 


5-O-desosamine of 14-membered macrolide antibiotics are important pharmacophores that bind with the target bacterial ribosome [1]. Direct modification of 5-O-desosamine was focused on the 2'–OH and 3'–NMe2 groups, which showed no antibacterial activity or weak antibacterial activity, respectively [2]. Due to the complexity of the macrolide structure and synthetic difficulty, there are few reports on the modification of other positions of desosamine [3]. In order to study other positions of 5-O-desosamine, one strategy is to synthesize new desosamine-mimetics to replace desosamine. Liang et al. synthesized several different 5-O-desosamine modified ketolides, in which 5-O-(6'-OBz)-desosamine ketolide showed excellent activities against several erythromycin-resistant pathogens [4, 5]. However, only the benzoyl group was studied for C-6 of desosamine. The method for C-6 modified desosamine and 5-O-(6'-O-modified)-desosamine ketolide was not published in detail. It would be meaningful to develop this synthetic method to study the 5-O-(6'-O-modified)-desosamine ketolides further. A series of 5-O-(4'-O-modified)-desosamine 14-membered ketolides [6] and 5-O-(4', 6'-O-dimodified)- desosamine 14-membered ketolides [7] were synthesized in our lab. Herein we report a new procedure of modifying C-6 of desosamine-mimetics and synthesizing a series of novel 5-O-(6'-O-modified)-desosamine 14-membered ketolides which could provide an opportunity for the design of a new class of ketolides.
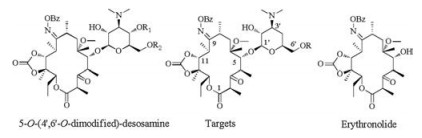
The targets (Fig. 1) adopted the strategy of replacement 5-O-desosamine with a modified desosamine mimetic. Erythronolide (Fig. 1) was obtained via 9 steps, using clarithromycin as the starting material [6, 7]. From a synthetic perspective, one challenge was to obtain suitable 6-O-modified desosamine donors to couple with erythronolide. In our previous work [7], the 4, 6-O-dimodified desosamine donors was synthesized and successfully coupled with erythronolide. After several steps, the result was 5-O-(4', 6'-O-dimodified)-desosamine ketolides.

|
Download:
|
| Fig. 1. The structures of 5-O-(4', 6'-O-dimodified)-desosamine ketolides, target compounds and erythronolide. | |
{kind=link}
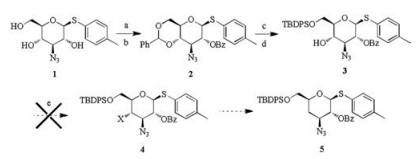
Therefore, the synthetic route for compound 5 was designed as shown in Scheme 1. 4-Methylphenyl-thiother was used as protective group for 1-position, which would be converted to trichloroacetimidate and used as a donor. The 3-NMe2 group was converted from the N3 group. The participation of neighboring group 2-OBz in the donors determined the glycosylation product of the β-configuration. Compound 1 was chosen as the starting material [7]. First, C4- and C6-dihydroxyl groups of compound 1 were protected by the benzal group with 4-methylbenzenesulfonic acid and PhCH(OMe)2 in a yield of 71%. Then, protection of the 2– OH group with benzoyl chloride (BzCl) formed compound 2 in 71% yield. The deprotection of compound 2 with trifluoromethanesulfonic acid (TFA) in water occurred readily. The 6-OH group was selectively protected with tert-butyldiphenylchlorosilane (TBDPSCl) in pyridine to form compound 3 in a yield of 76%. Deprotection of the TBDPS group occurred selectively under the condition of tetrabutylammonium fluoride (TBAF), which made the modification of C6-hydroxy possible. After the synthesis of compound 3, the deoxygenation reaction of C4-hydroxy was attempted.

|
Download:
|
| Scheme 1. Synthesis of compound 5. Reagents and conditions: (a) 4-methylbenzenesulfonic acid, PhCH(OMe)2, DMF, 71%; (b) BzCl, pyridine, 71%; (c) TFA, H2O, 94%; (d) TBDPSCl, DMAP, pyridine, 76%; (e) PPh3, CBr4 or I2, THF, failed. | |
{kind=link}
The Appel reaction [8-11] was used to replace OH with Br or I in the presence of PPh3, unfortunately, it did not work. The low activity of the secondary OH and the steric hindrance of the TBDPS group may be unfavorable for this conversion.
Therefore, the synthetic route was adjusted, as shown in Scheme S1 (Supporting information). The conversion of the C4- hydroxy group of compound 3 to C4-xanthate by the reaction with NaH/CS2/MeI in dry THF produced compound 6 in a yield of 86%. While Barton-McCombie deoxygenation was preceded with 2, 2'- azodiisobutyrodinitrile (AIBN) and n-Bu3SnH, an interesting result was observed. When 0.1 equiv. AIBN and 1.0 equiv. n-Bu3SnH were added to compound 6 in dry toluene and the mixture was heated to 50 ℃, most of compound 6 was transformed to a new compound 7. Alternatively, when 1.0 equiv. AIBN and 1.0 equiv. n-Bu3SnH were added to compound 6 in dry toluene and heated to 50 ℃, compound 6 was completely transformed to compound 8.
Compounds 7 and 8 were confirmed by 1H NMR, 13C NMR and HR-MS (Fig. S1 in Supporting information). By comparing the 1H NMR signals of compound 6 with compound 7, the signals for H-3 (3.96 ppm) and H-4 (6.06 ppm) of compound 6 were shifted to 6.04 ppm and 5.90 ppm (J = 10.0 Hz), which indicated that there is a C═C bond between C3 and C4. By comparing the 1H NMR signals of compound 6 with compound 8, the signals for H-4 (6.06 ppm) and H-2 (5.20 ppm) of compound 6 also shifted. The signal for H-4 (6.06 ppm) shifted upfield to 2.24 ppm and 1.72 ppm, which proved that the deoxygenation of C4-hydroxy was successful. In addition, the signal for H-2 (5.20 ppm) shifted upfield to 3.32 ppm and the NH (6.20 ppm) was identified, which showed that the 3-azido group of compound 6 was reduced to a 3-amino group and the 2– OBz group was converted to a 3–NHBz group.
When azido and xanthate were in the ortho-position, they could be transformed into different functional groups via radical reaction, by controlling the molar equivalents of AIBN (Fig. S1). It has been reported that the azido group participates in the reaction of free radicals upon free radical deoxygenation [12-17]. Aiming at avoiding free radical reactions, the azido group was adjusted. Triphenylphosphine was chosen to reduce the azido group of compound 6 by Staudinger reaction. The result showed that Chugaev elimination of compound 6 proceeded, and compound 9 was obtained (Scheme S2 in Supporting information).
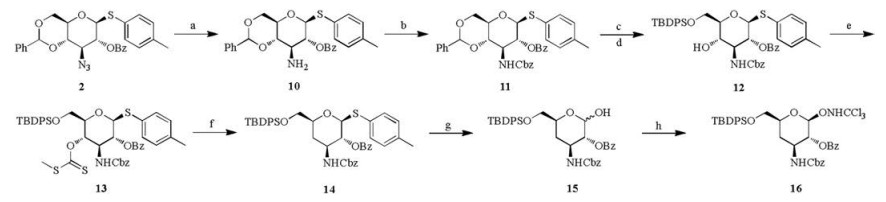
Based on the above result, the synthetic route was adjusted as shown in Scheme 2. The 6-benzyl-4-deoxy donor 16 was first synthesized from intermediate 2 via 8 steps. Staudinger reduction of the 3-azido group and protection of the 3-amino group with carbobenzoxy chloride (CbzCl) led to the formation of compound 11 in a yield of 89% in two steps. compound 12 was prepared following a similar procedure as compound 3. The 4–OH group of compound 12 was replaced with hydrogen via Barton-McCombie deoxygenation [18], and compound 14 was obtained in a yield of 39% in two steps. Then, hydrolysis of thioglycoside 14 with NIS/TFA formed compound 15 in DCM/H2O in a yield of 88% [19]. Donor 16 was produced by the reaction of compound 15 with CCl3CN and DBU in a yield of 74% [20].

|
Download:
|
| Scheme 2. Synthesis of compound 16. Reagents and conditions: (a) Ph3P, THF, H2O; (b) CbzCl, Na2CO3 aq., DCM, 89% for two steps; (c) TFA, DCM, H2O, 88%; (d) TBDPSCl, DMAP, pyridine, 78%; (e) CS2, MeI, NaH, THF, 79%; (f) AIBN, n-Bu3SnH, 90%; (g) NIS, TFA, DCM; (h) DBU, CCl3CN, DCM, 74% for two steps. | |
{kind=link}
Used TMSOTf as a promoter, erythronolide was successfully glycosylated with donor 16 to afford the glycosylated product 17 in a yield of 65%. The structure was confirmed by 1H NMR, 13C NMR and HR-MS spectra (H-1': 4.70 ppm, J = 7.2 Hz, β-configuration; C-1': 100.9 ppm; HR-MS [M + H]+: 1197.5295) (Scheme S3 in Supporting information). The reaction conditions were screened for removing the Cbz group (Table S1 in Supporting information). Under the condition of 1 equiv. of Pd(OH)2 with H2 (1.5–2 atm) over 4 h, monitored by TLC, the Cbz group was removed to give compound 18, and then methylation of the 30–NH2 group afforded compound 19 in 71% yield in two steps. The 3'–NMe2 group was confirmed by 1H NMR (3.37 ppm, s, 6H). We tried to remove the TBDPS group of compound 19 by TBAF, HF·Py or HCl/MeOH, but no target compound was obtained. We speculated that the macrolide ring might be unstable under those conditions.
The model reaction was carried that TBDPS protective group of compound 14 was removed by TBAF smoothly. Therefore, the synthetic route was adjusted, as shown in Scheme S4 (Supporting information). Levulinic (Lev) and Ac were chosen to replace the TBDPS group to protect the 6–OH group. Donors 22a and 22b were obtained by following similar procedure as that of compound 16.
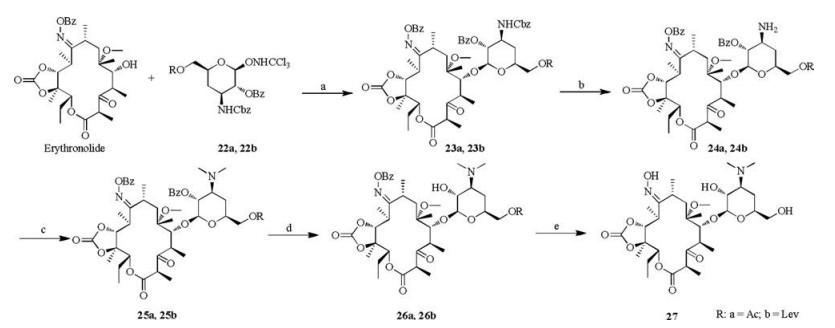
Target molecules 26a, 26b and 27 were prepared according to Scheme 3, following a similar procedure as that of compound 20. The difference, however, is that it is not necessary to remove the Ac and Lev protective groups, and target compounds 26a and 26b were obtained. The reaction time was prolonged to 48 h to give 27, a product that lacked the entire benzoyl protective group.

|
Download:
|
| Scheme 3. Synthesis of compound 27. Reagents and conditions: (a) TMSOTf, DCM, 63% for 23a; 44% for 23b; (b) Pd(OH)2, MeOH, 1.5–2.0 atm H2; (c) MeI, DMF, Na2CO3, 68% for 25a, 61% for 25b for two steps; (d) MeOH, reflux, 5 h, 80% for 26a; 7 h, 75% for 26b; (e) MeOH, reflux, 48 h, 71%. | |
{kind=link}
In order to obtain more products, Scheme S5 (Supporting information) outlines another attempt. The Lev protective group of compound 25b was treated with hydrazine hydrate to give the key intermediate compound 28. The unprotected 6'–OH group of compound 28 was modified in many ways, including the addition of ester, ether, and carbamate functional groups. Herein, a model reaction was carried out that modified the 6'–OH group with a benzoyl group, following deprotection in methanol to easily give compound 29. The result provides an opportunity to obtain various kinds of products from key intermediate 28 for better understanding the SARs of desosamine.
In conclusion, a procedure was developed for the synthesis of 5- O-(6'-O-modified)-desosamine 14-membered ketolides by adjustment of protective strategies and glycosylation conditions. Some novel ketolides were successfully synthesized, and the ketolides had different types of substituent groups at C-6' of desosamine mimetics. This work offers the opportunity to design more novel 5- O-(6'-O-modified)-desosamine-mimetics ketolides and contributes to a better understanding of the SARs of desosamine.
AcknowledgmentsThis work was supported by the CAMS Innovation Fund for Medical Sciences (Nos. 2017-I2M-3-011 and 2017-I2M-1-012). And we are grateful to the Department of Instrumental Analysis of our institute for performing the NMR and mass spectroscopy measurements.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2018.04.014.
| [1] |
(a) H. Takashima, Curr. Top. Med. Chem. 3 (2003) 991-999.
|
| [2] |
(a) N. LeTourneau, P. Vimal, D. Klepacki, A. Mankin, A. Melman, Bioorg. Med. Chem. Lett. 22 (2012) 4575-4578.
|
| [3] |
L.T. Phan, T. Jian, Z. Chen, et al., J. Med. Chem. 47 (2004) 2965-2968. DOI:10.1021/jm034233n |
| [4] |
A. Romero, C.H. Liang, Y.H. Chiu, et al., Tetrahedron Lett. 46 (2005) 1483-1487. DOI:10.1016/j.tetlet.2005.01.023 |
| [5] |
C.H. Liang, S. Yao, Y.H. Chiu, et al., Bioorg. Med. Chem. Lett. 15 (2005) 1307-1310. DOI:10.1016/j.bmcl.2005.01.027 |
| [6] |
(a) X.Z. Chen, P. Xu, Y.P. Xu, et al., Bioorg. Med. Chem. Lett. 22(2012) 7402-7405.
|
| [7] |
A.P. Wang, C. Liu, S. Yang, Z.H. Zhao, P.S. Lei, Tetrahedron 72 (2016) 285-297. DOI:10.1016/j.tet.2015.11.029 |
| [8] |
H. Arita, N. Ueda, Y. Matsushima, B Chem. Soc. JPN. 45 (1972) 567-569. DOI:10.1246/bcsj.45.567 |
| [9] |
T. Yoshisuke, N. Tomoko, K. Fumiyuki, K. Yukihiko, Heterocycle 44 (1997) 427-433. DOI:10.3987/COM-96-S41 |
| [10] |
W.L. Chen, C.F. Xia, J.H. Wang, et al., J. Org. Chem. 72 (2007) 9914-9923. DOI:10.1021/jo701539k |
| [11] |
P.D. Alexander, N.B. Charles, A.J.F. Michael, S.B. John, Tetrahedron Lett. 42 (2001) 117-119. DOI:10.1016/S0040-4039(00)01900-6 |
| [12] |
J. Xue, J. Wang, Z. Guo, Org. Lett. 6 (2004) 1365-1368. DOI:10.1021/ol0499046 |
| [13] |
P.B. Alper, S.C. Hung, C.H. Wong, Tetrahedron Lett. 37 (1996) 6029-6032. DOI:10.1016/0040-4039(96)01307-X |
| [14] |
K. Rakesh, I.W. Leonard, E.K. Edward, J. Med. Chem. 36 (1993) 2470-2474. DOI:10.1021/jm00069a004 |
| [15] |
B. Luisa, N. Daniele, S. Corrado, S. Piero, J. Org. Chem. 64 (1996) 7836-7841. |
| [16] |
E.F.V. Scriven, K. Turnbull, Chem. Rev. 88 (1988) 298-360. |
| [17] |
B. Lusia, L. Rino, M. Matteo, et al., Tetrahedron 58 (2002) 3485-3492. DOI:10.1016/S0040-4020(02)00302-2 |
| [18] |
A. Shazia, D.V. Natasha, E.R. Joseph, R.J.P. David, Tetrahedron 69 (2013) 816-825. |
| [19] |
J. Zhang, Y. Sun, X. Zhang, C. Li, H. Guan, Carbohydr. Res. 381 (2013) 74-82. DOI:10.1016/j.carres.2013.08.010 |
| [20] |
D. Mercedes, D.M. Julio, J. Org. Chem. 64 (1999) 4798-4810. DOI:10.1021/jo9901438 |