 2019, Vol. 30
2019, Vol. 30 


b Beijing Key Laboratory of Drug Resistance Tuberculosis Research, Department of Pharmacology, Beijing Tuberculosis and Thoracic Tumor Research Institute, Beijing Chest Hospital, Capital Medical University, Beijing 101149, China;
c Division of Gastroenterology and Hepatology, Department of Medicine, Indiana University School of Medicine, Indianapolis 46202, United States
Tuberculosis (TB) has existed for millennia and remains a major global health problem [1]. It is a widespread infectious disease predominantly caused by Mycobacterium tuberculosis (MTB), which can be transmitted through the air as droplets and affects the lungs [2]. The World Health Organization (WHO) estimated that approximately 10.4 million people were infected and 1.3 million died from TB worldwide in 2016 [1]. The spread of multidrugresistant TB (MDR-TB) and the emergence of extensively drugresistant TB (XDR-TB) have reinvigorated drug discovery efforts in search of novel agents [3-6]. Despite the introduction of Bedaquiline [7] and Delamanid [8] to the repertoire of anti-TB therapies for MDRTB, some adverse events have been noted [9]. Therefore, it is urgently needed to develop antimycobacterial molecules with new mechanisms of action and that are active against MDR-and XDR-TB [10].
Decaprenyl phosphoryl-β-D-ribose 2´-epimerase (DprE1) was identified as a potential target for developing potent and safer antiTB agents [11-13]. Some new chemical entities (NCEs) were found to have potent activity against MDR/XDR-MTB as covalent or noncovalent inhibitors of the DprE1 enzyme [14-22], such as nitroaromatic compounds DNB1, MTX and PBTZ 169(Fig. S1 in Supporting information). As the most advanced scaffold among these NCEs, nitrobenzothiazinones (BTZs) have garnered great interest recently, and many series of BTZ derivatives were reported [23-26]. Above all, candidate PBTZ169 entered in Phase Ⅱ clinical trials in 2017 [1].
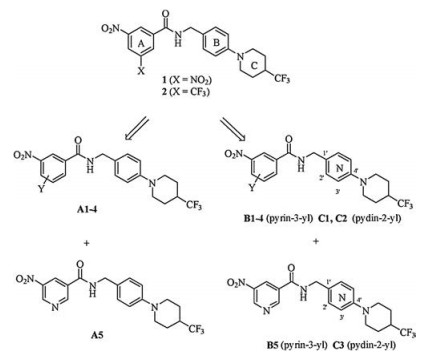
In our previous studies, many BTZs containing various cyclic ketoximes, spiro-heterocycles and piperidines moieties were found to have considerable antitubercular activity [27-29]. Recently, N-(4-(4-trifluoromethyl) piperidin-1-yl) benzyl nitrobenzamides 1 and 2(Fig. 1) were identified as new anti-TB agents by the thiazinone ring opening of PBTZ169 in our lab [30]. Both of them with simpler structures than PBTZ169, show potent activity against MTB H37Rv strain (MIC < 0.016 μg/mL). Moreover, compound 1 also displays acceptable safety and better PK properties than PBTZ169.
Inspired by the above research results, compounds 1 and 2 were employed as lead compounds, and the three moieties (A, BandC ring) were all explored in this study. We started with the modification of A ring and B ring. Replacement of X group on ring A with various substituents (Y) leaded to 3-nitrobenzamides bearing N-benzyl (A1-4); introduction of pyridine as A ring while reserving the nitro group gave 5-nitronicotinamides A5. Subsequently, the B ring was changed to pyrin-3-yl or pyrin-2-yl leading to compounds B1-5 or C1-3(Fig. 1). After identifying the optimal A and B rings, C ring was then further investigated. Our primary objective was to find optimized benzamides with potent antimycobacterial activity. A preliminary structure-activity relationship (SAR) study was also explored to facilitate the further development of these compounds.

|
Download:
|
| Fig. 1. Design of the new molecules. | |
Detailed synthetic pathways to side chains 6-8, leads 1, 2 and targets A-C are shown in Schemes S1 and S2(Supporting information), respectively. Commercially unavailable benzylamines and pyridinylmethylamines 6-8 were first prepared according to Scheme S1.4-Fluorobenzonitrile 3, 6-fluoronicotinonitrile 4 and 5-fluoropicolinonitrile 5 were treated with various nitrogen heterocyclic amines ZH in DMSO in the presence of K2CO3 at 80℃, and the resulting condensates were subsequently reduced with LiAlH4 in THF to produce the desired compounds 6, 7 and 8, respectively.
Leads 1, 2 and targets A1-11, B1-21, C1-4 were easily obtained by coupling 3-nitrobenzoic acids 9-13 and 5-nitronicotinic acid 14 with the above side chain compounds 6-8 or commercially available benzylamines 15a-d in the presence of triethylamine and condensation agent bis (2-oxo-3-oxazolidinyl) phosphonic chloride (BOP-Cl)(Scheme S2).
The target compounds A1-5, B1-5 and C1-3 bearing different kinds of substituents to ensure A and B rings flexibility and structure diversity, were first synthesized. They were preliminarily screened for in vitro activity against MTB H37Rv ATCC27294 strain, using the Microplate Alamar Blue Assay (MABA) [31, 32]. The minimum inhibitory concentration (MIC) is defined as the lowest concentration effecting a reduction in fluorescence of >90% relative to the mean of replicate bacterium-only controls. The MIC values of the compounds along with the leads 1 and 2, PBTZ169, isoniazid (INH), and rifampicin (RFP) for comparison were obtained from three independent experiments and presented in μg/mL in Table 1.
|
|
Table 1 Structures and activity of compounds A-C against MTB H37Rv. |
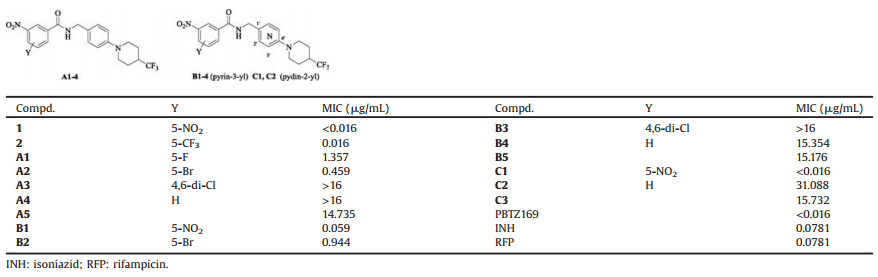
Effect of the substituents on A ring was first investigated. The nature and position of the substituents greatly influence activity. Replacement of one nitro group of 1 or the trifluoromethyl of 2 with halogen in compounds A1(F) and A2(Br) leads to decreased activity (MIC:1.357 and 0.459 μg/mL, respectively). Introduction of 4, 6-dichloro (A3) or reservation of one nitro (A4) destroys activity. Moreover, N-benzyl nicotinamide analogue (A5) displays very poor potency. Overall, these results reveal that the presence of a strong electron-withdrawing group (CF3, NO2) at C-5 position of nitrobenzamide core is essential for excellent activity (Table 1).
In further modifications, the benzene ring (B ring) was replaced by a pyridine ring. As shown in Table 1, in accordance with SAR of N-benzyl analogues (A1-5), N-(pyridin-3-yl) methyl and N-(pyridin-2-yl) methyl 3, 5-dinitrobenzamides (B1, C1) demonstrate potent MIC values of 0.059 and < 0.016 μg/mL against this strain, respectively, indicating that N-pyridinylmethyl on the amide is also acceptable.
Based on the above SAR, and better activity of lead compound 1 than 2, N-benzyl and N-pyridinylmethyl 3, 5-dinitrobenzamides with various groups at para-position of B ring were further designed and synthesized. As shown in Table 2, all of them show good to excellent activity against MTB H37Rv strain (MIC: < 0.016- 0.973 μg/mL), with one exception B15. Among them, nine compounds A6, 7, 9-11, B7, 8, 10 and C4(MIC: < 0.016-0.060 μg/mL) are more active than INH/RFP (MIC:0.0781 μg/mL), and roughly comparable to PBTZ169.
|
|
Table 2 Structures and activity of 3, 5-dinitrobenzamides A-C against MTB H37Rv. |

For N-benzyl 3, 5-dinitrobenzamides, the presence of a halogen atom instead of trifluoromethyl at para-position of the piperidine ring (C ring) was found to be also favorable. For example, compound A6 shows the same MIC value of < 0.016 μg/mL as the lead 1. Introduction of an additional aromatic moiety on C ring, such as 4-fluorophenyl (A7, MIC:0.059 μg/mL), is also acceptable. More interestingly, removal of C ring and direct attachment of a simple group to B ring remain considerable activity (A8-11), and an electron-donating group (OCH3) is preferred over an electronwithdrawing one (CF3, OCF3) or a halogen atom (F).
For N-(pyridin-3-yl) methyl 3, 5-dinitrobenzamides, the presence of a halogen atom (Cl, Br) instead of trifluoromethyl on C ring is more beneficial to activity (B1 vs. B7 and B8), and replacement of C ring in B1 with thiomorpholine in compound B10 maintains the same potent activity (MIC:0.059 μg/mL). However, introduction of 4-substituented phenyls on C ring, or replacement of the piperidine with piperazines bearing a substituted phenyl moiety leads decreased activity (B1 vs. B11-21). Conversely, N-(pyridin-2-yl) methyl compound C4 with a 4-(fluorophenyl) piperazine as C ring, displays the same potent MIC value of < 0.016 μg/mL as C1, much more active than the corresponding N-(pyridin-3-yl) methyl analogue B16(MIC:0.210 μg/mL)(Table 2).
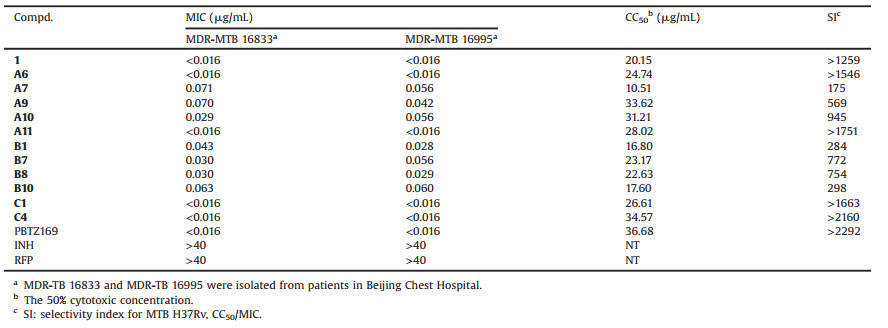
Encouraged by their strong potency against the drug sensitive MTB H37Rv strain (MIC: < 0.016-0.060 μg/mL), eleven 3, 5- dinitrobenzamide derivatives A6, 7, 9-11, B1, 7, 8, 10 and C1, 4 were further evaluated against two clinical isolated MTB-MDR (16, 833 and 16, 995) strains resistant to both INH and RFP. The cytotoxic potential of these compounds was also investigated in a mammalian Vero cell line by MTS assay. As shown in Table 3, all of them exhibit potent MIC values of < 0.016-0.071 μg/mL, similar to that against MTB H37Rv. Among of them, compounds A6, A11, C1 and C4 have the same excellent activity (MIC: < 0.016 μg/mL) as PBTZ169 and the lead 1. With a few exceptions, these compounds (CC50:22.63-34.57 μg/mL) are less cytotoxic than the lead 1, although generally more cytotoxic than PBTZ169.
|
|
Table 3 Activity against MDR-MTB, cytotoxicity and selectivity index (SI) values for selected compounds. |
{kind=link}
Lipinski's rules are important guidelines for determining drug-likeness compounds [33]. The related values of most potent compounds A6, A11, C1 and C4 were calculated using the online chemo-informatics software molinspiration (http://www. molinspiration. com). As shown in Table S1(Supporting information), none violation of Lipinski's rule-of-five was found among compounds A6, A11, and C1. The hydrogen bond acceptors of compound C4(HBA=11) are more than the recommended number (HBA < 10). However, compound C4 is still incorporate with the Lipinski's rule-of-five (violations < 1). Thus, these compounds display good drug like properties, are all deserved further development.
In conclusion, a series of nitrobenzamide derivatives containing N-benzyl or N-pyridinylmethyl moieties, based on lead compounds 1 and 2 discovered in our lab, were designed and synthesized as new anti-TB agents. Many of them exhibit potent in vitro antitubercular activity. Especially, N-benzyl 3, 5-dinitrobenzamides A6 and A11, and N-(pyridine-2-yl) methyl analogues C1 and C4 have not only the same excellent activity (MIC: < 0.016 μg/ mL) against both drug-sensitive MTB strain H37Rv and two drugresistant clinical isolates as PBTZ169 and the lead 1, but also have acceptable safety indices (SI:>1500). In addition, compounds A6, A11, C1 and C4 display good drug like properties, suggesting these compounds may serve as new and promising candidates for further antitubercular drug discovery. By the way, the further expansion of the 3, 5-dinitrobenzamides is underway to find potent anti-TB agents.
AcknowledgementsThis work is supported by the National Mega-project for Innovative Drugs (Nos. 2015ZX09102007-008, 2015ZX09102007- 015, 2015ZX0 9304006-016, 2018ZX09721001-004-007, 2018ZX09711001-007-002), CAMS Initiative for Innovative Medicine (No. 2016-I2M-1-010), CAMS Innovation Fund for Medical Science (Nos. CAMS-2016-I2M-1-010, CAMS-2017-I2M-1-011), PUMC Youth Fund (No. 2017350011).
Appendix A.Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.08.005.
| [1] |
Global Tuberculosis Report, World Health Organization, 2017, www.who.Int/tb/publications/lpa-mdr-diagnostics/en/
|
| [2] |
N. Nayak, J. Ramprasad, U. Dalimba, et al., Chin. Chem. Lett. 27 (2016) 365-369. DOI:10.1016/j.cclet.2016.01.015 |
| [3] |
A. Wang, Y. Yang, J. Yang, et al., Bioorg. Med. Chem. 26 (2018) 2073-2084. DOI:10.1016/j.bmc.2018.03.004 |
| [4] |
Z. Xu, S. Zhang, C. Gao, et al., Chin. Chem. Lett. 28 (2017) 159-167. DOI:10.1016/j.cclet.2016.07.032 |
| [5] |
K. Lv, L. Li, B. Wang, et al., Eur. J. Med. Chem. 137 (2017) 117-125. DOI:10.1016/j.ejmech.2017.05.044 |
| [6] |
R.S. Wallis, M. Maeurer, P. Mwaba, et al., Lancet Infect. Dis. 16 (2016) 34-46. DOI:10.1016/S1473-3099(16)00070-0 |
| [7] |
A.K. Kakkar, N. Dahiya, Tuberculosis (Edinb.) 94 (2014) 357-362. DOI:10.1016/j.tube.2014.04.001 |
| [8] |
M.T. Gler, V. Skripconoka, E. Sanchez-Garavito, et al., N. Engl. J. Med. 366 (2012) 2151-2160. DOI:10.1056/NEJMoa1112433 |
| [9] |
D.T. Hoagland, J. Liu, R.B. Lee, R.E. Lee, Adv. Drug Deliv. Rev. 102 (2016) 55-72. DOI:10.1016/j.addr.2016.04.026 |
| [10] |
Beena, D.S. Rawat, Med. Res. Rev. 33 (2013) 693-764. DOI:10.1002/med.2013.33.issue-4 |
| [11] |
G. Manina, M.R. Pasca, S. Buroni, E. de Rossi, G. Riccardi, Curr. Med. Chem. 17 (2010) 3099-3108. DOI:10.2174/092986710791959693 |
| [12] |
G. Riccardi, M.R. Pasca, L.R. Chiarelli, et al., Appl. Microbiol. Biotechnol. 97 (2013) 8841-8848. DOI:10.1007/s00253-013-5218-x |
| [13] |
M. Brecik, I. Centarova, R. Mukherjee, et al., ACS Chem. Biol. 10 (2015) 1631-1636. DOI:10.1021/acschembio.5b00237 |
| [14] |
P.K. Crellin, R. Brammananth, R.L. Coppel, PloS One 6 (2011) e16869. DOI:10.1371/journal.pone.0016869 |
| [15] |
S.A. Stanley, S.S. Grant, T. Kawate, et al., ACS Chem. Biol. 7 (2012) 1377-1384. DOI:10.1021/cb300151m |
| [16] |
F. Wang, D. Sambandan, R. Halder, et al., Proc. Natl. Acad. Sci. U. S. A. 110 (2013) 2510-2517. DOI:10.1073/pnas.1309171110 |
| [17] |
M. Naik, V. Humnabadkar, S.J. Tantry, et al., J. Med. Chem. 57 (2014) 5419-5434. DOI:10.1021/jm5005978 |
| [18] |
M. Panda, S. Ramachandran, V. Ramachandran, et al., J. Med. Chem. 57 (2014) 4761-4771. DOI:10.1021/jm5002937 |
| [19] |
P.S. Shirude, R. Shandil, C. Sadler, et al., J. Med. Chem. 56 (2013) 9701-9708. DOI:10.1021/jm401382v |
| [20] |
T. Christophe, M. Jackson, H.K. Jeon, et al., PLoS Pathog 5 (2009) e100645. |
| [21] |
K.J. Schaper, M. Pickert, A.W. Frahm, Archiv der Pharmazie 332 (1999) 91-102. |
| [22] |
C. Trefzer, M. Rengifo-Gonzalez, M.J. Hinner, et al., J. Am. Chem. Soc. 132 (2010) 13663-13665. DOI:10.1021/ja106357w |
| [23] |
V. Makarov, G. Manina, K. Mikusova, et al., Science 324 (2009) 801-804. DOI:10.1126/science.1171583 |
| [24] |
V. Makarov, B. Lechartier, M. Zhang, et al., EMBO Mol. Med. 6 (2014) 372-383. DOI:10.1002/emmm.201303575 |
| [25] |
A.L.D.L. Ribeiro, G. Degiacomi, F. Ewann, et al., PloS One 6 (2011) e26675. DOI:10.1371/journal.pone.0026675 |
| [26] |
T. Karoli, B. Becker, J. Zuegg, et al., J. Med. Chem. 55 (2012) 7940-7944. DOI:10.1021/jm3008882 |
| [27] |
R. Zhang, K. Lv, B. Wang, et al., RSC Adv. 7 (2017) 1480-1483. DOI:10.1039/C6RA25712G |
| [28] |
K. Lv, X. You, B. Wang, et al., ACS Med. Chem. Lett. 8 (2017) 636-641. DOI:10.1021/acsmedchemlett.7b00106 |
| [29] |
K. Lv, Z. Tao, Q. Liu, et al., Eur. J. Med. Chem. 151 (2018) 1-8. DOI:10.1016/j.ejmech.2018.03.060 |
| [30] |
L. Li, K. Lv, Y. Yang, et al., ACS Med. Chem. Lett. 9 (2018) 741-745. DOI:10.1021/acsmedchemlett.8b00177 |
| [31] |
L. Collins, S.G. Franzblau, Antimicrob. Agents Chemother. 41 (1997) 1004-1009. DOI:10.1128/AAC.41.5.1004 |
| [32] |
Y. Lu, M. Zheng, B. Wang, etal., Antimicrob. AgentsChemother. 55 (2011) 5185-5193. DOI:10.1128/AAC.00699-11 |
| [33] |
C.A. Lipinski, F. Lombardo, B.W. Dominy, P.J. Feeney, Adv. Drug Deliv. Rev. 46 (2001) 3-26. DOI:10.1016/S0169-409X(00)00129-0 |