 2019, Vol. 30
2019, Vol. 30 


The use of phenol derivatives instead of aryl halides as aryl electrophiles in cross-coupling reactions has received considerable attention in recent years [1-9]. They are generally cost-effective, readily available and environmentally friendly since no halogencontaining waste is generated during the coupling reactions. Among the phenol derivatives, substrates bearing 2-pyridyloxy group (OPy) showed wide applications in C-H activation reactions due to the strong coordination ability of the nitrogen atom and their chemical stability [10]. A very important feature of such phenol derivatives lies in that the Py or OPy group can be selectively removed after C-H functionalization, thereby facilitating the formation of high value-added products. For example, the Py group could be removed in MeOTf/MeONa system [11]. Our group achieved a palladium-catalysed regioselective C-H acetoxylation of 2-aryloxypyridines [12]. After removal of the Py group, a series of poly-substituted phenols were obtained. Recently, the removal of OPy group has also been disclosed. Chatani et al. developed a rhodium [13] or nickel [14] catalysed borylation of 2- aryloxypyridines, providing a facile approach to introduce multiple functional groups into an aromatic ring. Wang et al. reported a nickel-catalysed amination of 2-aryloxypyridines after initial C-H activations [15] (Scheme 1a).

|
Download:
|
| Scheme 1. Applications of phenol derivatives through C-O bond cleavage. | |
Despite these significant progress, the reductive deoxygenation of phenol derivatives has received less attention. It was worth noting that reductive deoxygenation of phenol derivatives is also of significant importance in synthetic chemistry, enabling the use of oxygen-based auxiliaries as removable directing groups [16]. It can also be applied to synthesize deoxygenated analogs of phenolbased natural products [17]. During the past decades, phenol mesylates, triflates, tosylates, sulfonates, and even aryl alkyl ethers were successfully reduced in the presence of transition metal catalysts as well as the reductants [18]. Just recently, Wang et al. [19] reported a nickel-catalysed C-O bond reduction of aryl and benzyl 2-pyridyl ethers using simple alkoxide [20] as the reductant. However, most of these reactions were carried out in a hazardous solvent such as DMF and toluene under harsh reaction conditions.
2-MeTHF is recognized as an eco-friendly solvent which can be obtained from renewable resources. It is also an attractive solvent in organometallic reactions due to its stronger Lewis basicity compared with THF and Et2O [21], which may facilitate its coordination to metal center. Moreover, 2-MeTHF is only partially miscible with water, making the separation process simplified. Inspired by these pioneering works, herein we report a nickelcatalysed reduction of 2, 4, 6-triaryloxy-1, 3, 5-triazines (TAT) in 2- MeTHF (Scheme 1b).
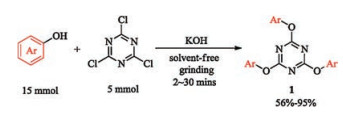
Initially, 2, 4, 6-triaryloxy-1, 3, 5-triazine 1 were chosen as the substrate since such compound could be readily prepared in the presence of a base. The phenols and TCT are also readily available and cost-effective. Moreover, the TAT structure is similar with 2- aryloxypyridine. Notably, all of the three aryl groups in TAT can be utilized [22]. To further simplify the preparation of substrate 1, we tried the grinding method under solvent-free condition. Pleasingly, most of the reactions proceeded smoothly to provide the corresponding products in moderate to excellent yields within 30 min (Scheme 2 and Supporting information). The pure products could be obtained by washing with water and hexane, and no further purification was required.

|
Download:
|
| Scheme 2. Preparation of 2, 4, 6-triaryloxy-1, 3, 5-triazines 1. | |
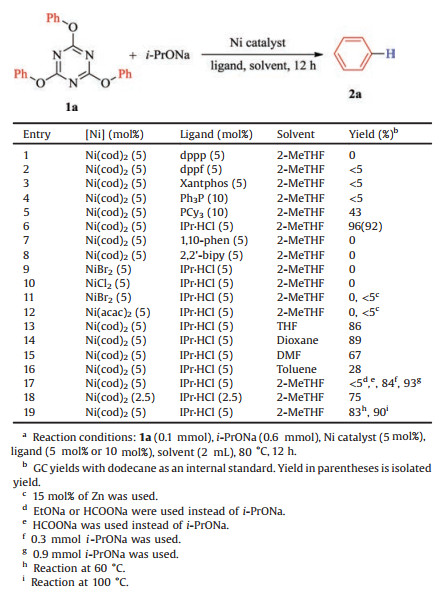
Then we started to explore the reductive deoxygenation reaction using 2, 4, 6-triphenoxy-1, 3, 5-triazine 1a as the model substrate and i-PrONa as the reductant in2-MeTHFat80℃ (Table 1). The tentative reaction usingNi (cod)2 as the catalyst, 1, 3-bis (diphenylphosphino)- propane (dppp) as the ligand led to the failure of the reaction (entry 1). Switching the ligands to 1, 1'-bis (diphenylphosphino) ferrocene (dppf), 4, 5-bis (diphenylphosphino)-9, 9-dimethylxanthene (Xantphos), and Ph3P afforded the desired products in only trace amount (< 5%, entries 2-4). The addition of PCy3, which was reported as the most the effective ligand [18c-e,18g]for reductive deoxygenation reactions, provided the desired product in 43% yield (entry 5). Pleasingly, when the reaction was performed by employing 1, 3-bis- (2, 6-diisopropylphenyl) imidazolinium chloride (IPr·HCl) as the ligand, the yield increased up to 92%(entry 6). Some bidentate nitrogen ligands were also checked, but no reaction occurred at all (entries 7 and 8). Different nickel precatalysts such as NiBr2 and Ni (acac)2 were also tried, however, no reaction was observed. Only trace amount of the desired products were detected in the presence of zinc (entries 9 and 10). The solvent also had a significant influence on the reaction. Comparable yields were obtained when the reaction took place either in THF or dioxane (entries 13 and 14). Reaction in other solvents such as DMF and toluene only gave 67% and 28% yields, respectively (entries 15 and 16). Other reductants such as EtONa and HCOONa were also evaluated, however, only trace amount of desired product was observed. Reduce the amount of i-PrONa to 1 equiv. (relative to phenyl group) resulted in a slightly decreasedyield (84%). No further increase in the yield was not iced in the presence of 3 equiv. of i-PrONa (entry 17). Finally, attempt reaction with lower catalyst loading (2. 5 mol%) also gave a lower yield (75%), and 80℃ was turned out to be the optimum.
|
|
Table 1 Optimization of reaction conditions.a |
{kind=link}
{kind=link}
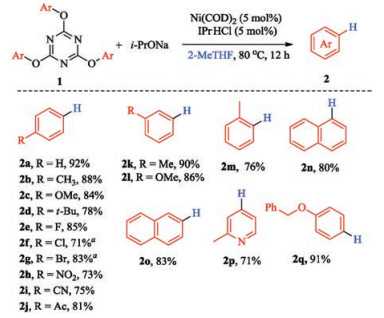
With the optimized conditions in hand, a series of 2, 4, 6- triaryloxy-1, 3, 5-triazines were utilized to explore the scope of this protocol (Fig. 1). Generally, all of the investigated substrates underwent the reductive deoxygenation reaction smoothly to provide the corresponding products in moderate to excellent yields (71%-92%). Among para-substituted substrates, diverse functional groups including alkyl, methoxyl, fluoro, nitro, cyano, and acyl were all well tolerated. However, substrates with chloro and bromo group (2f and 2g) did not survive the reaction conditions, and dehalogenation products followed by C-O bond reduction were obtained as the major products. The metasubstituted substrates also exhibited good reactivities to deliver the corresponding products 2k and 2l in excellent yields. Importantly, although ortho-methyl substituted substrate 1m gave relatively lower yield which was probably due to the hindrance effect, the yield was still satisfactory. Compared with 2-naphthol TAT substrate, relatively lower yield was obtained for 1-naphthol TAT substrate due to the steric hindrance. This protocol is also applicable for heterocyclic substrates, giving the corresponding product 2p in 71% yield. For substrate 1q, 91% yield was obtained. The benzyloxy group which can be easily reduced by catalytic hydrogenolysis, was kept intact under the present reaction conditions.

|
Download:
|
| Fig. 1. The scope of this protocol. Reaction conditions:1(0.1 mmol), i-PrONa (0.6 mmol), Ni (COD)2(5 mol%), IPr·HCl (5 mol%), 2-MeTHF (2 mL), 80℃, 12 h. Isolated yields. aDehalogenation product followed by C-O reduction was obtained as the major product. | |
{kind=link}
Scale-up experiment was also performed using compound 1s as the model substrate (5 mmol scale, Scheme 3). This reaction also proceeded smoothly to provide the desired product 2q in 83% yield, indicating the practicability of the present protocol.

|
Download:
|
| Scheme 3. Scale-up experiment. | |
{kind=link}
Since the 2, 4, 6-triaryloxy-1, 3, 5-triazines have similar structure with 2-aryloxypyridines, the good coordination ability of the sp2 nitrogen atoms can facilitate a number of ortho C-H functionalization reactions. After C-O reduction, a series of substituted arenes could be obtained. Thus, sequential C-H functionalization/ C-O reduction reactions were carried out (Scheme 4). For example, the Pd-catalyzed arylation [10b] and Ru-catalyzed oxidative olefination [23] could introduce an ortho aryl and vinyl, respectively, on the aromatic ring. After C-O bond reduction, products 3 and 4 were obtained in moderate yields. Notably, the ester group in compound 4 was not affected under this reaction conditions.

|
Download:
|
| Scheme 4. Sequential C–H functionalization/C–O bond reduction. | |
{kind=link}
Deuterium experiment was then performed to gain preliminary mechanistic information about this reaction (Scheme 5 and Supporting information). When the reaction was carried out with a deuterated sodium isopropoxide, the corresponding deuterated product 2b-D was obtained in 82% yield with 96% deuteration, indicating that sodium isopropoxide was the hydrogen donor of the reduction reaction. Also, acetone was detected after the reduction reaction by GC-MS.

|
Download:
|
| Scheme 5. Deuterium experiment. | |
{kind=link}
A plausible reaction mechanism was also proposed in Scheme 6. Initially, the nickel catalyst coordinates with ortho nitrogen atom of substrate 1, then C-O oxidative addition of 1 to Ni (0) catalyst gives intermediate A [22]. Subsequently, intermediate C is generated via anion exchange with i-PrO-, which releases acetone and intermediate D. Finally, intermediate D undergoes reductive elimination to give the desired product 2 with simultaneous regeneration of Ni (0) species.

|
Download:
|
| Scheme 6. Proposed mechanism. | |
{kind=link}
In summary, we have developed a nickel-catalysed reduction of phenol derivatives activated by 2, 4, 6-trichloro-1, 3, 5-triazine in ecofriendly 2-MeTHF. The phenol-TCT derivatives were readily prepared using grinding method in a short time without further purification. Caryl-O bond was selectively cleavaged while no products formed via CPy-O bond cleavage were observed. The reactions proceeded under mild reaction conditions with high efficiency and good functional group tolerance. Gram-scale reaction was also achieved on a 5 mmol scale. Particularly, the success of sequential C-H functionalization of phenol-TCT derivatives followed by C-O bond reduction provided a new way to access functionalized arenes.
AcknowledgmentsWe gratefully acknowledge financial support from the National Natural Science Foundation of China (Nos. 21302014 and 21676030), the Jiangsu Key Laboratory of Advanced Catalytic Materials and Technology (No. BM2012110), the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions, the Advanced Catalysis and Green Manufacturing Collaborative Innovation Center of Changzhou University, and Topnotch Academic Programs Project of Jiangsu Higher Education Institutions (No. PPZY2015B145).
| [1] |
B.M. Rosen, K.W. Quasdorf, D.A. Wilson, et al., Chem. Rev. 111 (2011) 1346-1416. DOI:10.1021/cr100259t |
| [2] |
B. Li, D. Yu, C. Sun, Z. Shi, Chem. Eur. J. 17 (2011) 1728-1759. DOI:10.1002/chem.201002273 |
| [3] |
T. Mesganaw, N.K. Garg, Org. Process Res. Dev. 17 (2013) 29-39. DOI:10.1021/op300236f |
| [4] |
J. Cornella, C. Zarate, R. Martin, Chem. Soc. Rev. 43 (2014) 8081-8097. DOI:10.1039/C4CS00206G |
| [5] |
M. Tobisu, N. Chatani, Acc. Chem. Res. 48 (2015) 1717-1726. DOI:10.1021/acs.accounts.5b00051 |
| [6] |
X. Cong, H. Tang, X. Zeng, J. Am. Chem. Soc. 137 (2015) 14367-14372. DOI:10.1021/jacs.5b08621 |
| [7] |
W. Shi, X. Li, Z. Li, Z. Shi, Org. Chem. Front. 3 (2016) 375-379. DOI:10.1039/C5QO00395D |
| [8] |
J. Tang, M. Luo, X. Zeng, Synlett 28 (2017) 2577-2580. DOI:10.1055/s-0036-1588568 |
| [9] |
H. Zeng, Z. Qiu, A. Domínguez-Huerta, et al., ACS Catal. 7 (2017) 510-519. DOI:10.1021/acscatal.6b02964 |
| [10] |
(a) X. Jia, S. Zhang, W. Wang, F. Luo, J. Cheng, Org. Lett. 11 (2009) 3120-3123; (b) J. Chu, P. Lin, M. Wu, Organometallics 29 (2010) 4058-4065; (c) L. Ackermann, E. Diers, A. Manvar, Org. Lett. 14 (2012) 1154-1157; (d) M. Kim, S. Sharma, J. Park, et al., Tetrahedron 69 (2013) 6552-6559; (e) J. Yao, R. Feng, Z. Wu, Z. Liu, Y. Zhang, Adv. Synth. Catal. 355 (2013) 1517-1522; (f) B. Liu, H. Jiang, B. Shi, Org. Biomol. Chem. 12 (2014) 2538-2542; (g) N. Khatun, A. Banerjee, S. K. Santra, A. Behera, B. K. Patel, RSC Adv. 4 (2014) 54532-54538; (h) B. Liu, H. Jiang, B. Shi, J. Org. Chem. 79 (2014) 1521-1526; (i) W. Zhang, J. Zhang, S. Ren, Y. Liu, J. Org. Chem. 79 (2014) 11508-11516; (j) C. Zhang, P. Sun, J. Org. Chem. 79 (2014) 8457-8461; (k) Y. Xu, P. Liu, S. Li, P. Sun, J. Org. Chem. 80 (2015) 1269-1274; (l) M. K. Raghuvanshi, K. Rauch, L. Ackermann, Chem. Eur. J. 21 (2015) 1790-1794; (m) Y. Liang, X. Li, X. Wang, et al., ACS Catal. 5 (2015) 1956-1963; (n) J. Chu, S. Chen, M. Chiang, M. Wu, Organometallics 34 (2015) 953-966; (o) H. Lou, Q. Chen, Y. Wang, et al., ACS Catal. 5 (2015) 2846-2849. |
| [11] |
H. Xu, K. Muto, J. Yamaguchi, et al., J. Am. Chem. Soc. 136 (2014) 14834-14844. DOI:10.1021/ja5071174 |
| [12] |
L. Wang, L. Pan, Y. Huang, Q. Chen, M. He, Eur. J. Org. Chem. 2016 (2016) 3113-3118. DOI:10.1002/ejoc.201600508 |
| [13] |
H. Kinuta, M. Tobisu, N. Chatani, J. Am. Chem. Soc. 137 (2015) 1593-1600. DOI:10.1021/ja511622e |
| [14] |
M. Tobisu, J. Zhao, H. Kinuta, et al., Adv. Synth. Catal. 358 (2016) 2417-2421. DOI:10.1002/adsc.201600336 |
| [15] |
J. Li, X. Wang, Org. Lett. 19 (2017) 3723-3726. DOI:10.1021/acs.orglett.7b01549 |
| [16] |
C.J. Rohbogner, G.C. Clososki, P. Knochel, Angew. Chem. Int. Ed. 47 (2008) 1503-1507. |
| [17] |
N.Z. Burns, I.N. Krylova, R.N. Hannoush, P.S. Baran, J. Am. Chem. Soc. 131 (2009) 9172-9173. DOI:10.1021/ja903745s |
| [18] |
(a) V. Kogan, Tetrahedron Lett. 47 (2006) 7515-7518; (b) B. H. Lipshutz, B. A. Frieman, T. Butler, V. Kogan, Angew. Chem. Int. Ed. 45 (2006) 800-803; (c) P. Alvarez-Bercedo, R. Martin, J. Am. Chem. Soc. 132 (2010) 17352-17353; (d) M. Tobisu, K. Yamakawa, T. Shimasaki, N. Chatani, Chem. Commun. 47 (2011) 2946-2948; (e) A. G. Sergeev, J. F. Hartwig, Science 332 (2011) 439-443; (f) A. G. Sergeev, J. D. Webb, J. F. Hartwig, J. Am. Chem. Soc. 134 (2012) 20226-20229; (g) J. Cornella, E. Gómez-Bengoa, R. Martin, J. Am. Chem. Soc. 135 (2013) 1997-2009; (h) Y. Ren, M. Yan, J. Wang, Z. C. Zhang, K. Yao, Angew. Chem. Int. Ed. 51 (2013) 12674-12678; (i) M. Tobisu, T. Morioka, A. Ohtsuki, N. Chatani, Chem. Sci. 6 (2015) 3410-3414; (j) A. Ohgi, Y. Nakao, Chem. Lett. 45 (2016) 45-47; (k) E. M. Wiensch, D. P. Todd, J. Montgomery, ACS Catal. 7 (2017) 5568-5571; (l) K. Yasui, M. Higashino, N. Chatani, M. Tobisu, Synlett 28 (2017) 2569-2572. |
| [19] |
J. Li, Z. Wang, Chem. Commun. 54 (2018) 2138-2141. DOI:10.1039/C7CC09668B |
| [20] |
(a) C. J. E. Davies, M. J. Page, C. E. Ellul, M. F. Mahon, M. K. Whittlesey, Chem. Commun. 46 (2010) 5151-5153; (b) Y. Yi, W. Yang, D. Zhai, et al., Chem. Commun. 52 (2016) 10894-10897. |
| [21] |
(a) D. F. Aycock, Org. Process Res. Dev. 11 (2007) 156-159; (b) V. Pace, P. Hoyos, L. Castoldi, P. D. de Maria, A. R. Alcantara, ChemSusChem 5 (2012) 1369-1379; (c) Y. Gu, F. Jerome, Chem. Soc. Rev. 42 (2013) 9550-9570; (d) A. Farran, C. Cai, M. Sandoval, et al., Chem. Rev. 115 (2015) 6811-6853; (e) R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sadaba, M. Lopez Granados, Energy Environ. Sci. 9 (2016) 1144-1189. |
| [22] |
(a) N. Iranpoor, F. Panahi, Adv. Synth. Catal. 356 (2014) 3067-3073; (b) N. Iranpoor, F. Panahi, Org. Lett. 17 (2015) 214-217; (c) N. Iranpoor, F. Panahi, F. Jamedi, J. Organomet. Chem. 781 (2015) 6-10; (d) E. Etemadi-Davan, N. Iranpoor, Chem. Commun. 53 (2017) 12794-12797. |
| [23] |
W. Ma, L. Ackermann, Chem. Eur. J. 19 (2013) 13925-13928. DOI:10.1002/chem.201301988 |