 2019, Vol. 30
2019, Vol. 30 


b Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300072, China
Converting carbon dioxide (CO2) into economic feedstock chemical or fuels has become more and more important because of the rising of CO2 levels in the atmosphere. Generally, a variety of production could be derived from CO2 [1] and one of the most important products, formic acid (HCOOH) can be employed as both important feedstock's and a potential hydrogen storage material within the"hydrogen energy economy" [2]. A multitude of homogeneous catalysts mainly concentrated on Ru, Ir, Rh complex have been prepared and applied for CO2 hydrogenation to HCOOH [3]. However, the difficulty of separation and recovery of homogeneous catalysts hinder the process of industrialization of CO2 hydrogenation to formic acid. Thus, it is imperative to develop heterogeneous catalyst to catalyze the formation of formic acid.
Ru-based homogeneous catalysts have been extensively studied in the synthesis of formic acid due to their favorable activity [4], but few research about heterogeneous Ru-based catalyst were reported previously. Our group developed Ru(OH)x/γ-AL2O3 by precipitation method as the active catalyst for CO2 hydrogenation to formic acid [5], and Ru(OH)x/γ-AL2O3 gave a TOF of 139 h-1 under supercritical CO2 conditions. We also prepared the RuCl3-DBU/γ-AL2O3 catalyst, giving the TOF of 239 h-1, under 15MPa and 80 ℃ at the presence of solvent dimethyl sulfoxide (DMSO), trimethylamine(Et3N), and protonic additive KH2PO4 [6]. 'Si'-(CH2)3-NH(CSCH3)-RuCl3-PPh3 catalyst was prepared by Zhang et al. [7] and TOF of 103 h-1 was obtained in ionic liquid under 18MPa (CO2/H2=1:1) at 60℃. Yu et al. [8] developed a catalyst, which consisted of functionalized mesoporous MCM-41 and anchored ruthenium complex, achieving a TOF of 1022 h-1, under 14. 7 MPa (CO2/H2=9.3:5.4) at 80℃. Thus, the exploration of Ru complex supported catalysts for CO2 hydrogenation to formic was still relatively rare and had enormous potential to develop an effective Ru-based catalyst.
Metal-organic-frameworks (MOFs) are a kind of porous materials, and it is worth noting that MOFs are composed of inorganic second-structure unit (SBU) and organic linkers [9]. Organic-inorganic hybrid structure of MOFs materials makes them outstanding features including flexible pore size, high specific surface, large porosity and thermos stability. MOFs are widely applied for gas storage and separation, catalysis, even functional materials areas such as optics, electricity and magnetism [10]. The organic linkers of containing amino group of the MOFs can be functionalized and attached to transition metal ions by postsynthetic modification (PSM) [11], which could produce more versatile MOF-based materials with various physical and chemical properties. MIL-101(Cr)-NH2 was widely studied as a good candidate of PSM because of relatively large pores, favorable thermal stability as well as excellent chemical stability to water and some organic solvents [12]. Wang et al. [13] prepared Co(Ⅱ)@Cr-MIL-101-P2I catalyst by introducing pyridine-3-aldehyde to the amino group of MIL-101(Cr)-NH2, and applied it as heterogeneous catalysts for aerobic epoxidation of olefins.
In this paper, a series of novel metal-organic frameworkanchored RuCl3 catalyst for the CO2 hydrogenation to formic acid have been developed. RuCl3 was immobilized onto mesoporous MIL-101(Cr)-NH2 using salicylaldehyde, and DPPBde as linker agents. The amino group of MIL-101(Cr)-NH2 produced Schiff-base moieties in the MOF skeleton by condensation with aldehyde group of linker agents. The Schiff-base moieties can play a role of bidentate ligand and anchor the Ru(Ⅲ)ions as active site of catalyst. The high surface area of MIL-101(Cr)-NH2 is favorable for dispersion of Ru(Ⅲ)ions. The promoting effect of solvents and additives were also explored and to elevate the catalytic performance of CO2 hydrogenation to formic acid.
MIL-101(Cr)-NH2 was synthesized according to the hydrothermal synthesis method with a modification reported by the literature [14]. MIL-101(Cr)-Sal was prepared by the reaction between MIL-101(Cr)-NH2 with salicylaldehyde [13], and the final product was labeled as MIL-101(Cr)-Sal. The MIL-101(Cr)-DPPB was prepared by the similar procedure as MIL-101(Cr)-Sal but with DPPBde instead of salicylaldehyde. RuCl3@MIL-101(Cr)-Sal was prepared following the process that the methanol solution of MIL- 101(Cr)-Sal and RuCl3·xH2O refluxed under stirring at 85℃ for 6 h, and the last solid production was labeled as RuCl3@MIL-101(Cr)- Sal. The preparation of RuCl3@MIL-101(Cr)-DPPB and RuCl3@MIL- 101(Cr)-NH2 were the same as RuCl3@MIL-101(Cr)-Sal, but the support materials were replaced with MIL-101(Cr)-DPPB and MIL- 101(Cr)-NH2. The detailed synthetic conditions were deposited in Supporting information.
For a typical procedure of catalyst evaluation, 20 mg of the catalyst, 30 mL of solvent, and 15 mL of Et3N were added into a 100 mL stainless steel autoclave with a magnetic stirrer. After flushing the autoclave four times with CO2, CO2/H2 in a certain proportion was filled into autoclave up to the desired operating pressure. The autoclave was heated to target temperature by mantle equipped with a temperature controller. After reacting for 2 h, the reactor was cooled to room temperature in ice water bath before depressurizing. Then, the liquid product was examined by high performance liquid chromatography. Turnover numbers (TON) was calculated to reflect the yield of formic acid.
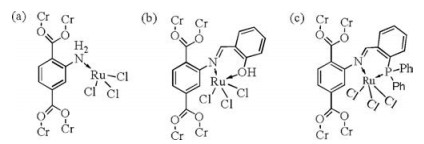
The possible existing coordination structure of Ru(Ⅲ)in different RuCl3@MOF catalyst are shown in Fig. 1. In Fig. 1a, RuCl3 is directly coordinated with the N atom of the amino group in the MIL-101(Cr)-NH2, which may exist in three different catalysts. In Fig. 1b, RuCl3 coordinates with the N atom of carbon nitrogen double bonds and the O atom of the phenolic hydroxyl from salicylaldehyde, and this coordination form exists only in the RuCl3@MIL-101(Cr)-Sal. In Fig. 1c, RuCl3 coordinates with the N atom of carbon nitrogen double bonds and the P atom from DPPBde and this coordination form exists only in the RuCl3@MIL-101(Cr)- DPPB. The catalytic performances of the different catalyst for CO2 hydrogenation to formic acid were shown in Table 1. The reaction conditions were as following:The mass of catalysts labeled as mc was 20 mg, initial total pressure labeled as Pt0 was 6 MPa, the temperature was 80℃, and the reaction time labeled as tr was 2h. Obviously, the pure carrier has no activity for the synthesis of formic acid. RuCl3 as catalyst obtained relatively poor activity compared with RuCl3@MOF catalysts. The amino group and Schiff base moieties in MOF skeleton can coordinate with Ru(Ⅲ)ions and act as electron donors for Ru(Ⅲ)ions, which further promoted the formation of formate intermediate through accelerating the nucleophilic attack of the Ru-H intermediate ligand to CO2 [15]. RuCl3@MIL-101(Cr)-DPPB gave the best performance for producing formic acid than RuCl3@MIL-101(Cr)-NH2 and RuCl3@MIL-101(Cr)- Sal, which may be attributed to stronger coordination interaction of bidentate ligands originated from DPPBde with Ru(Ⅲ)center.

|
Download:
|
| Fig. 1. The possible coordination forms of RuCl3 in RuCl3@MOF. | |
|
|
Table 1 The catalytic performance of the different catalysts for CO2 hydrogenation to formic acida |

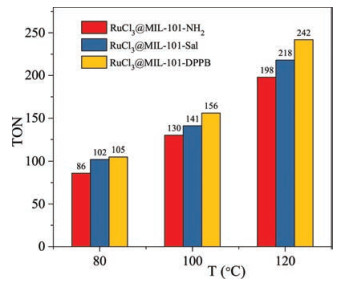
Because of the low activity of RuCl3@MOF catalysts at 80℃, the catalytic performance was further explored at higher temperatures, and the TON of formic acids is shown in Fig. 2. It is noted that the reactivity of three catalysts increased with the rising of the temperature. RuCl3@MIL-101(Cr)-NDPPB gave the highest TON of 242 at 120℃, 6 MPa.

|
Download:
|
| Fig. 2. Catalytic performance of the different catalysts with the different temperatures. | |
Further, the effects of the solvent and additive on the CO2 hydrogenation to formic acid were explored with RuCl3@MIL-101(Cr)- DPPB as model catalyst at 120℃ and the results are listed in Table 2. Several organic polar solvents were employed to reveal their effects in the production of formic acid. TON of formic acid was 278, 242, 198 with methanol, ethanol, and isopropanol as solvent, respectively. The higher TON of 406 was obtained in non-proton strong polar dimethyl sulfoxide (DMSO) solvent. Ohnishi et al. [15] clarified that the polar solvent promoted the insertion of CO2 to the RuH bond, which was generally considered as the rate-determining step of CO2 hydrogenation to formic acid. The polarity of these solvents were in the orderof DMSO > methanol > ethanol > isopropanol and the order of catalytic performance of catalyst in different solvents was positive correlation with the polar order of the solvent.
|
|
Table 2 Catalytic performance of CO2 hydrogenation to formic acid in different solventsa |

{kind=link}
{kind=link}
It was noteworthy that the miscibility between Et3N and DMSO was very poor. H2O molecular can improve the mutual solubility of solution system and promote the absorption of CO2 in solution [16, 17]. TON of formic acid was improved to 664 by adding 5 mL of water to replace part of DMSO in Et3N and DMSO solution. In addition, H2O molecular is a proton type solvent and can serve as a hydrogen-bond donor [18] or a strong ligand to promote reaction [19]. PPh3 as free ligands can replace solvent molecules and coordinate with Ru(Ⅲ)catalytic centers as a donor ligand in the reaction of CO2 hydrogenation to formic acid, and further enhanced the reaction activity by providing electrons to the metal center. When 3mmol PPh3 was added, the reactivity was improved significantly, representing optimal 831 of TON of formic acid over RuCl3@MIL-101 (Cr)-DPPB with DMSO and H2O as themixed solvents and Et3N as the base additive under 6 MPa, 120℃, reaction time of 2 h.
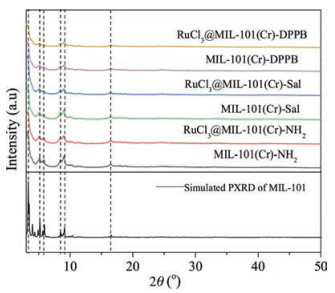
The XRD patterns of three MOFs and the corresponding RuCl3@MOF catalysts are shown in Fig. 3. It is noted that the XRD patterns of the as-synthesized MIL-101(Cr)-NH2 were well consistent with the simulated XRD patterns, which indicated that the MIL-101(Cr)-NH2 materials were successfully synthesized. The MIL-101(Cr)-Sal, RuCl3@MIL-101(Cr)-Sal, MIL-101(Cr)-DPPB and RuCl3@MIL-101(Cr)-DPPB showed the diffraction patterns similar with MIL-101(Cr)-NH2, meaning that the framework structure of MIL-101(Cr)-NH2 remained relatively intact after PSM and RuCl3 loading. The XRD patterns of RuCl3 was not observed, demonstrating high dispersion of Ru(Ⅲ)in MOFs. The absence of XRD patterns of Ru metal and RuO2 indicated that Ru(Ⅲ)species are stable in the process of preparation, further confirming that the active centers of catalysts were contributed to Ru(Ⅲ)species.

|
Download:
|
| Fig. 3. Powder XRD patterns of the different MOF carriers and RuCl3@MOF catalysts. | |
{kind=link}
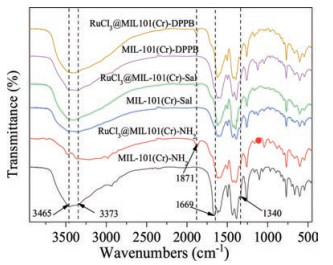
As illustrated in Fig. 4, FT-IR spectra were employed to prove the effective formation of the covalent functionalized compound. The double peaks at 3475 cm-1 and 3382 cm-1 of MIL-101(Cr)-NH2 were attributed to the asymmetrical and symmetrical stretching vibration adsorption of the amino groups. Besides, the peak at 1654 cm-1 can be ascribed to the N-H bending vibration of primary amino groups, and the characteristic bands of 1340 cm-1 is for the stretching vibration of C-N in benzene ring [20]. After PSM and anchoring of RuCl3, the relative intensities of peaks at 3475 cm-1 and 3382 cm-1 was decreased obviously and the characteristic bands of the N-H bending vibration at 1654 cm-1 disappeared, which indicated the formation of the C=N bond [14]. However, the bond of C=N of the Schiff-base was not well-resolved due to the overlapping with the bending vibration band of the primary amino groups [21]. It was worth noting that FT-IR spectra of RuCl3@MOF after anchoring of RuCl3 showed a new band at 1887 cm-1, which can be relationship with the introduction of RuCl3 [22].

|
Download:
|
| Fig. 4. FT-IR spectra of different MOF carriers and RuCl3@MOF catalysts. | |
{kind=link}
The pore structure and specific surface area of all the samples are evaluated using N2 physical adsorption measurement and the adsorption isotherms are shown in Fig. S1 in Supporting information. All the samples featured the same type IV isotherm, which demonstrated that the pore structure was well maintained after PSM of the linker and anchoring of RuCl3. As shown in Table S1 in Supporting information, the specific surface area of the samples was calculated by using Brunauer-Emmett-Teller (BET) method and Langmuir method, separately. The BET (Langmuir) surface area and the total pore volume of the MIL-101(Cr)-NH2 sample were 2322(3851)m2/g and 1.21 m3/g, respectively. An obvious decrease in the BET (Langmuir) surface area and the total pore volume was observed for the samples of PSM and anchoring RuCl3, which indicated that the pores of MIL-101(Cr)-NH2 were occupied by Ru(Ⅲ)complexes. The RuCl3@Cr-MIL-101(Cr)-DPPB sample had the lowest BET surface area and total pore volume, which could be ascribed to the bigger molecular volume of DPPBde.
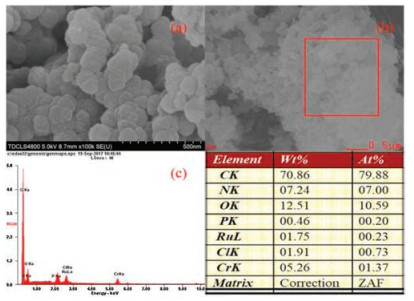
Typical morphologies of MIL-101(Cr)-NH2, MIL-101(Cr)-101-Sal and MIL-101(Cr)-DPPB are shown in Fig. S2 in Supporting information. SEM image of MIL-101(Cr)-NH2(Fig. S2a)showed that the as-synthesized MOF was constituted of particles with the size around 20-100 nm, which was the same as the previous reports [14]. As shown in Fig. S3 in Supporting information, MIL- 101(Cr)-NH2 was spheroidal particles, which was not damaged after PSM with salicylaldehyde or DPPBde. The phosphorus(P) element was introduced to RuCl3@MIL-101(Cr)-DPPB in the PSM process, and the SEM-EDX results of RuCl3@MIL-101(Cr)-DPPB shown in Figs. 5b and c confirmed the presence of Ru, Cl and P. Furthermore, the atomic ratio of Ru/Cl/P on the RuCl3@MIL-101 (Cr)-DPPB was close to 1:3:1, which indicated that RuCl3 coordinated with P derived from DPPBde in the form of Fig. 1c. TEM images of RuCl3@MIL-101(Cr)-NH2, RuCl3@MIL-101(Cr)-Sal, and RuCl3@MIL-101(Cr)-DPPB are shown in Fig. 6. It can be seen from Fig. 6 that three RuCl3@MOF catalysts were all composed of particles less than 50 nm. The aggregated Ru particles were not observed, meaning that the high dispersion of RuCl3. For RuCl3@MIL-101(Cr)-DPPB, the best active catalyst, the elements distribution of Ru, Cl and P on the catalyst were further explored by TEM-EDX-mapping shown in Fig. 6d. The results indicated that Ru, Cl, and P evenly distributed throughout the surface of the MOF.

|
Download:
|
| Fig. 5. (a) SEM image and(b, c)SEM-EDS analysis of RuCl3@MIL-101(Cr)-DPPB. | |
{kind=link}

|
Download:
|
| Fig. 6. TEM images of RuCl3@MIL-101(Cr)-NH2(a), MIL-101(Cr)-Sal(b), RuCl3@MIL- 101(Cr)-DPPB(c)and EDS-mapping of RuCl3@MIL-101(Cr)-DPPB(d). | |
{kind=link}
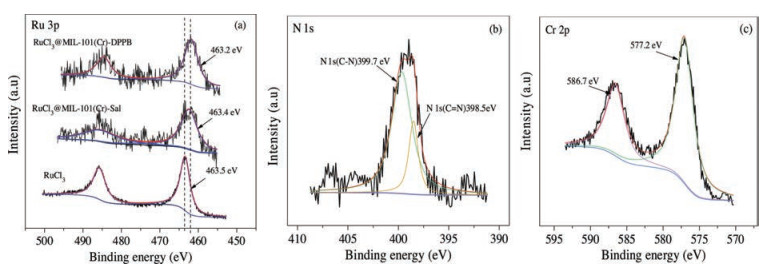
In order to gain thorough insight into the surface state of the catalysts, the XPS spectrum of the RuCl3@MIL-101(Cr)-DPPB sample was investigated. As shown in Fig. 7c, the Cr 2p1/2 and Cr 2p3/2 signals consisted of two peaks at 586.7 eV and 577.2 eV, respectively, which corresponded to the typical binding energies of Cr3+ [23]. The peak-fitted N 1 s core-line spectrum of RuCl3@MIL- 101(Cr)-DPPB(Fig. 7b)showed two resolved peaks at 399. 7 eV and 398.5 eV, corresponding to imine(C=N)groups [24], amino(-NH2) groups [25] respectively, which had also proved the achievement of the PSM of MIL-101(Cr)-NH2. For RuCl3@MIL-101(Cr)-DPPB, the binding energy of Ru 3p5/2 core level peak was at 462.2 eV(Fig. 7a), which shifted towards a lower value compared with RuCl3 at 483.5 eV and RuCl3@MIL-101(Cr)-Sal at 462.4 eV. Lower binding energy of Ru 3p in RuCl3@MIL-101(Cr)-DPPB indicated that the DPPB ligand had stronger electron donating ability for Ru center, which caused higher activity for CO2 hydrogenation to formic acid.

|
Download:
|
| Fig. 7. (a) The Ru 3p XPS peaks of the different catalysts; (b)N 1s XPS peaks of RuCl3@MIL-101(Cr)-DPPB; (c)Cr 2p XPS peaks of RuCl3@MIL-101(Cr)-DPPB. | |
{kind=link}
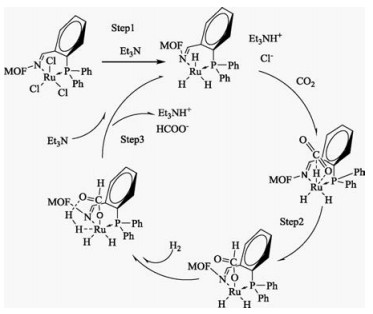
Reaction mechanism of hydrogenation of CO2 to formic acid by RuCl3@MIL-101(Cr)-DPPB catalyst was proposed in Scheme 1, which based on Yuya Ohnishi's report in homogeneous catalytic system [15]. The Step 1 was the formation process of Ru-H bond. The Step 2 was CO2 inserting Ru-H bond. The Step 3 was the process of H2 activation which consists of the coordination of dihydrogen molecule with the Ru center, the isomerization of the Ru(Ⅱ)η1- formate intermediate, and the metathesis. The rate determining step was the process of CO2 inserting into Ru-H and stronger donation ability could decrease the activation barrier of the insertion of CO2 into the Ru(Ⅱ)-H bond. For RuCl3@MIL-101(Cr)- NH2, RuCl3@MIL-101(Cr)-Sal, RuCl3@MIL-101(Cr)-DPPB catalysts, it was noted that the ligand in the different MOF had the different donation ability, as shown in Ru3p XPS spectrum. MIL-101(Cr)- DPPB had the strongest donation ability among three catalysts, thus resulting in the best activity for CO2 hydrogenation to formic acid. Furthermore, the PPh3 is a common kind of electron-rich ligand and can dramatically improve the reactivity of CO2 hydrogenation to formic acid. According to Scheme 1, it can be known that CO2 was activated by Ru-H bond which acted as actual active center and then inserted into Ru-H bond to form formate intermediate. The addition of electron-rich ligand on metals would decrease the Lewis acidity of the catalyst, but it would also change the strength of Ru-H bond which is favorable for the process of CO2 inserting into Ru-H. Thus, the reactivity was increased when free PPh3 as electron-rich ligand were used due to the facilitation of CO2 inserting Ru-H bond process.

|
Download:
|
| Scheme 1. Reaction mechanism of hydrogenation of CO2 to formic acid by RuCl3@MIL-101(Cr)-DPPB. | |
{kind=link}
In summary, several novel heterogeneous RuCl3@MOF catalysts were synthesized by PSM of MIL-101(Cr)-NH2 and anchoring RuCl3, and applied for CO2 hydrogenation to formic acid. Characterized by a combination of FT-IR, XRD, TG, N2 physical adsorption, SEM, TEMEDX and XPS, it was demonstrated that, for RuCl3@MIL-101(Cr)- DPPB, bidentate ligand from DPPBde was successfully introduced into the MOF skeleton of MIL-101(Cr)-NH2, and Ru and P were coordinated and well dispersed on the MOF. RuCl3@MIL-101(Cr)- DPPB catalyst exhibited the higher catalytic performance for hydrogenation of CO2 to formic acid than RuCl3@MIL-101(Cr)-NH2 and RuCl3@MIL-101(Cr)-Sal due to the P atom of DPPBde as a stronger electron-donor substituent to promote insertion of CO2 into RuH bond. Moreover, compared with methanol, ethanol and isopropanol, DMSO with stronger polarity was demonstrated to be a superior solvent to improve the reactivity of CO2 hydrogenation. H2O as proton additive can promote the absorption of CO2 in solution, improve the mutual solubility of solution system, and serve as a hydrogen-bond donor. PPh3 played a role as electron donor ligand to coordinate with Ru(Ⅲ)center and improved reactivity. TON of 831 was obtained for CO2 hydrogenation to formic acid over RuCl3@MIL-101(Cr)-DPPB under 6 MPa(CO2/ H2=1:1)at 120℃ with DMSO and H2O as the mixed solvents and Et3N as the base additive in the presence of PPh3. The development of the RuCl3@MIL-101(Cr)-DPPB solid catalysts brings more inspiration for the design of highly active heterogeneous catalyst of CO2 hydrogenation to formic acid.
AcknowledgmentsFinancial support from the National Natural Science Foundation of China(NSFC, Nos. 21776211, 21325626), the Program for New Century Excellent Talents in University(No. NCET-13-0411)and the Program of Introducing Talents of Discipline to Universities(No. B06006)is gratefully acknowledged.
Appendix A.Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.06.021.
| [1] |
Q. Liu, L. Wu, R. Jackstell, et al., Nat. Commun. 6 (2015) 5933-8947. DOI:10.1038/ncomms6933 |
| [2] |
M. Pérez-Fortes, J.C. Schöneberger, A. Boulamanti, et al., Int. J. Hydrogen Energy 41 (2016) 16444-16462. DOI:10.1016/j.ijhydene.2016.05.199 |
| [3] |
C. Federsel, R. Jackstell, M. Beller, Angew. Chem. Int. Ed. 49 (2010) 6254-6257. DOI:10.1002/anie.201000533 |
| [4] |
K. Sordakis, C. Tang, L.K. Vogt, et al., Chem. Rev. 118 (2018) 372-433. DOI:10.1021/acs.chemrev.7b00182 |
| [5] |
C. Hao, S. Wang, M. Li, et al., Catal. Today 160 (2011) 184-190. DOI:10.1016/j.cattod.2010.05.034 |
| [6] |
W. Zhang, S. Wang, Y. Zhao, et al., Chem. Lett. 45 (2016) 555-557. DOI:10.1246/cl.160013 |
| [7] |
Z. Zhang, Y. Xie, W. Li, et al., Angew. Chem. Int. Ed. 47 (2008) 1127-1129. |
| [8] |
Y.M. Yu, J.H. Fei, Y.P. Zhang, et al., Chin. J. Chem. 24 (2006) 840-844. |
| [9] |
J. Gascon, A. Corma, F. Kapteijn, et al., ACS Catal. 4 (2014) 361-378. DOI:10.1021/cs400959k |
| [10] |
Y. Cui, B. Li, H. He, et al., Accounts Chem.Res. 49 (2016) 483-493. DOI:10.1021/acs.accounts.5b00530 |
| [11] |
S.M. Cohen, Chem. Sci. 1 (2010) 32-36. DOI:10.1039/c0sc00127a |
| [12] |
Y. Huang, S. Liu, Z. Lin, et al., J. Catal. 292 (2012) 111-117. DOI:10.1016/j.jcat.2012.05.003 |
| [13] |
J. Wang, M. Yang, W. Dong, et al., Catal. Sci. Technol. 6 (2016) 161-168. DOI:10.1039/C5CY01099C |
| [14] |
Y. Lin, C. Kong, L. Chen, RSC. Adv. 2 (2012) 6417-6419. DOI:10.1039/c2ra20641b |
| [15] |
Y.Y. Ohnishi, T. Matsunaga, Y. Nakao, et al., J. Am. Chem. Soc. 127 (2005) 4021-4032. DOI:10.1021/ja043697n |
| [16] |
J. Benitez-Garcia, G. Ruiz-Ibanez, A. Bidarian, et al., Chem. Eng. Sci. 45 (1990) 3407-3415. DOI:10.1016/0009-2509(90)87146-J |
| [17] |
R.J. Littel, W.P.M.V. Swaaij, G.F. Versteeg, AIChE. J. 36 (1990) 1633-1640. |
| [18] |
C. Yin, Z. Xu, S.Y. Yang, et al., Organometallics 20 (2001) 1216-1222. DOI:10.1021/om000944x |
| [19] |
K. Rohmann, J. Kothe, M.W. Haenel, et al., Angew. Chem. Int. Ed. 55 (2016) 8966-8969. DOI:10.1002/anie.201603878 |
| [20] |
X. Li, Y. Pi, Q. Xia, et al., Appl. Catal. B: Environ. 191 (2016) 192-201. DOI:10.1016/j.apcatb.2016.03.034 |
| [21] |
D. Saha, R. Sen, T. Maity, et al., Langmuir 29 (2013) 3140-3151. DOI:10.1021/la304147j |
| [22] |
S. Wu, L. Chen, B. Yin, et al., Chem. Com. 51 (2015) 9884-9887. DOI:10.1039/C5CC02741A |
| [23] |
H. Khajavi, H.A. Stil, H.P.C.E. Kuipers, et al., ACS Catal. 3 (2013) 2617-2626. DOI:10.1021/cs400681s |
| [24] |
S. Bhattacharyya, C. Cardinaud, G. Turban, J. Appl. Phys. 83 (1998) 4491-4500. DOI:10.1063/1.367211 |
| [25] |
S. Bhattacharyya, J. Hong, G. Turban, J. Appl. Phys. 83 (1998) 3917-3919. DOI:10.1063/1.367312 |