 2019, Vol. 30
2019, Vol. 30 


b Department of Chemistry, University of South Florida, Tampa FL33620, United States
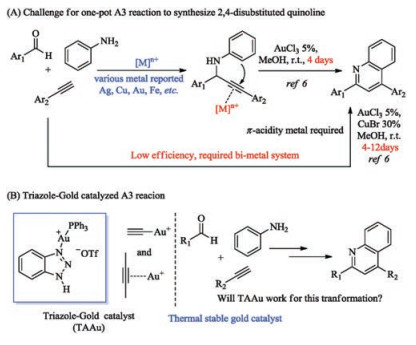
Multicomponent reactions have been raised tremendous attention in the past, as it provided efficient ways to construct complex molecules through a one-pot reaction [1, 2]. Among them, three components coupling reaction of aldehyde, alkyne and amine, known as A3 coupling reaction, is particularly interesting [3]. Generally, this protocol contains two sequential steps. The first step involved the addition of alkyne to imine, which was generated by aldehyde and amine in situ, to afford propargylic aryl amine derivatives. Various of metals with Lewis acid properties have been reported to promote this step, such as Ag, Cu, Fe, etc. [4, 5]. The second step involved the activation of internal alkyne. In the presence of aryl amine, an intramolecular electrophilic aromatic substitution to alkyne will take place to furnish the 2, 4- disubstituted quinoline derivatives under the oxidative conditions (Scheme 1A) [6]. Quinoline is one category of heterocyclic structures with interesting biologic activities [7, 8]. Therefore, A3 reaction is one of the valuable protocols to quick assemble quinoline motif. However, the difficulty of internal alkyne activation makes the cyclization step become challenging.

|
Download:
|
| Scheme 1. A3 Reaction. | |
Few metal catalysts have the capability to serve as both Lewis acid for the first step and π-acid for the internal alkyne activation. Therefore, a new metal catalyst to promote one-pot reaction is highly desired.
Gold catalysis has been flourished over the past decade [9, 10]. In particular, as an efficient carbophilic Lewis acid, gold (Ⅰ) catalysis has facilitated numerous interesting transformations through the activation of alkynes, allenes, and alkenes [11, 12]. Therefore, gold catalyst will be a good candidate for this transformation. As shown in Scheme 1A, Wang reported the first successful example to synthesis 2, 4-disubstituted quinoline from propargyl amine under gold catalysis conditions [6]. AuCl3 was found to be the optimal catalyst for cyclization of propargyl amine. However, the overall reaction efficiency was largely restricted by the stability of gold catalyst. At room temperature, it took days (4 days) to finish the cyclization. When increasing the temperature, AuCl3 will decompose quickly into gold mirror or nano-particles. Microwave assistant was demanded for a fast reaction kinetic (15 min), however, sacrificing the yield. Another drawback of AuCl3 is that it could not promote the addition of alkyne to imine. To achieve the one pot reaction, CuBr was employed for the alkyne addition. Moreover, the substrate scope is limited to 2, 4-diaryl-quinoline, due to the problematic internal alkyl alkyne activation. Recently, Lin and coworkers reported the synthesis of 4-alkyl-quinoline with the combination of CuCl and AuCl [13]. Notably, Cu salt is still required to form copper acetylene addition to imine. Since gold acetylene has already been widely reported, and the addition of gold acetylene to various electrophiles, such as oxonium cation, has been well investigated [14]. It is reasonable to be expected that A3 coupling reaction could be achieved with gold catalyst alone. One plausible reason is that Au (Ⅰ) cation is required for effective gold acetylene formation, which more tend to decompose at high temperature. Our group has a long interest in gold catalysis and demonstrated the application of triazole-Au complexes as thermally stable catalysts in challenging alkyne activation under harsh conditions, such as intermolecular alkyne hydroamination and Hashmi phenol synthesis [15, 16]. Triazole can form a dynamic coordination with Ph3PAu+cation, which allows the effective activation of internal alkyne with improved gold cation stability. Inspired by previous results, we are wondering if triazole-gold (TAAu) could be applied in this transformation (Scheme 1B).
With this in mind, our initial attempt was carried out using 1a, 2a, and 3a together with triazole-gold catalyst at room temperature (Table 1, entry 2). To our delight, the propargyl amine product was observed with 37% yield with incomplete conversion, indicating the feasible of gold catalyzed propargyl amine formation. When raising the temperature to 60 ℃, cyclization product 5a was started to form along with 36% of 4. This result suggested a higher temperature was required for internal alkyne activation. The reaction was completed with 90% yield of quinoline product 5a at 110 ℃ after 24 h. PPh3AuNTf2 and PPh3AuOTf was also applied but obtained decreased yield of 5a, mainly due to the decomposition of Au (Ⅰ) cation at a high temperature. Some gold (Ⅲ) catalysts were also tested and no desired products (either 4 or 5a) were detected, which suggested that gold acetylene was unable to be formed from Au (Ⅲ) cation. Notably, the decomposition of triazolegold (by formation of gold mirror) was not observed during the reaction, which highlighted the thermal stability of triazole-gold catalyst.
|
|
Table 1 Triazole-gold catalyzed A3 reaction. |

{kind=link}
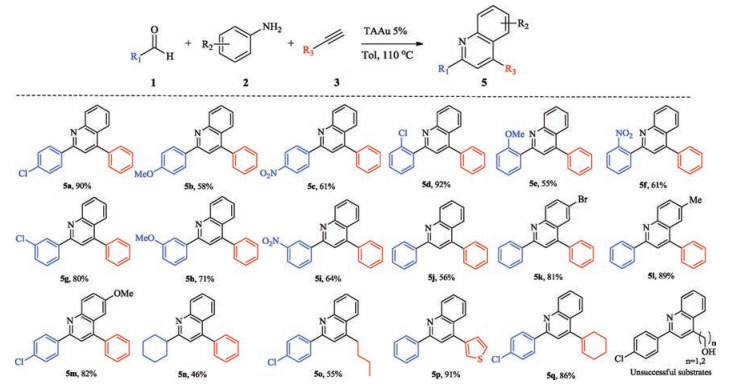
With this optimal condition in hand, we further explored the substrate scope as summarized in Fig. 1. Aryl aldehydes with different substituents (para, ortho, meta) were tested. Generally, electron deficient aryl aldehydes afforded better yields due to more feasible imine formation (5a, 5c). The position of substituents has no obvious effect (5c, 5f, 5i). Although benzaldehyde provided modest yield, switching to electro-rich aniline, p-toluidine (5l) and p-anisidine (5m), the yield was increased, indicating the second cyclization of internal alkyne is the rate limiting step. Besides aryl aldehydes, aliphatic aldehyde (5m) were also proved to be tolerable under this condition. Furthermore, more challenging alkyne substrates were also evaluated, such as 3-thienylacetlyene (5n), 1-hexyne (5o), and 1-ethynylcyclohexene (5q), revealing the excellent capability of TAAu towards internal alkyne activation. Both propargyl alcohol and homo propargyl alcohol failed to provide the desired products and only imine was observed. It might due to the inter/intra molecular cyclization of propargyl alcohol under TAAu catalysis condition, which diminished gold acetylene formation. Overall, the success of these substrates indicated the first example of one-pot A3 reaction under gold catalysis.

|
Download:
|
| Fig. 1. Substrate scopes. Standard condition:5 mol% TAAu, 1(1.1 mmol), 2(1.0 mmol), and 3(1.0 mmol) in toluene (1 mL). The reaction mixture was stirred at 110 ℃ for 24- 36 h. Isolated yield. | |
{kind=link}
In conclusion, we disclosed the triazole gold catalyzed A3 coupling reaction to synthesize 2, 4-disubstituted quinoline derivatives. With this protocol, simple and stable TAAu was used as catalyst alone for the overall sequential reaction, providing a broad substrate scope. Notably, 4-alkyl substituted, or 2-alkyl substituted quinoline derivatives were obtained with good yield, which highlighted the superior of our triazole-gold catalyst in the combination of both stability and reactivity.
AcknowledgmentsWe are grateful to the National Science Foundation (No. CHE- 1619590), NIH (No. 1R01GM120240-01) and the National Nature Science Foundation of China (No. 21629201) for financial support. We also thank the Development Project of the Pharmaceutical Industry of Jilin Province (Nos. 20150311070YY, 20170307024YY).
Appendix A.Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.05.036.
| [1] |
H. Bienaymé, C. Hulme, G. Oddon, P. Schmitt, Chem. Eur. J. 6 (2000) 3321-3329. |
| [2] |
L.F. Tietze, Chem. Rev. 96 (1996) 115-136. DOI:10.1021/cr950027e |
| [3] |
C. Wei, Z. Li, C.J. Li, Org. Lett. 5 (2003) 4473-4475. DOI:10.1021/ol035781y |
| [4] |
C.M. Wei, Z.G. Li, C.J. Li, Synlett (2004) 1472-1483. |
| [5] |
V.A. Peshkov, O.P. Pereshivko, E.V. Van der Eycken, Chem. Soc. Rev. 41 (2012) 3790-3807. DOI:10.1039/c2cs15356d |
| [6] |
F. Xiao, Y. Chen, Y. Liu, J. Wang, Tetrahedron 64 (2008) 2755-2761. DOI:10.1016/j.tet.2008.01.046 |
| [7] |
P.Y. Chung, Z.X. Bian, H.Y. Pun, et al., Future Med. Chem. 7 (2015) 947-967. DOI:10.4155/fmc.15.34 |
| [8] |
V.R. Solomon, H. Lee, Curr. Med. Chem. 18 (2011) 1488-1508. DOI:10.2174/092986711795328382 |
| [9] |
D.J. Gorin, F.D. Toste, Nature 446 (2007) 395-403. DOI:10.1038/nature05592 |
| [10] |
S. Sengupta, X.D. Shi, ChemCatChem 2 (2010) 609-619. DOI:10.1002/cctc.201000070 |
| [11] |
D. Pflasterer, A.S.K. Hashmi, Chem. Soc. Rev. 45 (2016) 1331-1367. DOI:10.1039/C5CS00721F |
| [12] |
A. Furstner, Chem. Soc. Rev. 38 (2009) 3208-3221. DOI:10.1039/b816696j |
| [13] |
K.M. Jiang, J.A. Kang, Y. Jin, J. Lin, Org. Chem. Front. 5 (2018) 434-441. DOI:10.1039/C7QO00637C |
| [14] |
S. Hosseyni, C.A. Smith, X. Shi, Org. Lett. 18 (2016) 6336-6339. DOI:10.1021/acs.orglett.6b03228 |
| [15] |
H.F. Duan, S. Sengupta, J.L. Petersen, N.G. Akhmedov, X.D. Shi, J. Am. Chem. Soc 131 (2009) 12100. DOI:10.1021/ja9041093 |
| [16] |
Y.F. Chen, W.M. Yan, N.G. Akhmedov, X.D. Shi, Org. Lett. 12 (2010) 344-347. DOI:10.1021/ol902680k |