 2019, Vol. 30
2019, Vol. 30 


Chirality is an omnipresent phenomenon in nature, manifested by natural products including nucleic acids, proteins, amino acids, and sugars [1, 2]. These materials exhibit specific activity and functionality which are attributed to their chirality. As a result, great efforts have been paid to prepare various kinds of chiral compounds and separate their enantiomers. Among them, chiral phthalocyanines have attracted growing interest in the last three decades owing to their extensive biological relevance and various potential applications [3-5]. Chiral phthalocyanines reported could be roughly divided into three groups:(i) phthalocyanines containing chiral carbons in the side chain; (ii) phthalocyanines with optically active aromatic molecules; (iii) phthalocyanines with planar asymmetry[5a,b]. More attention was given to the first ones owing to their self-assembly properties. To date, the studies have focused on introducing chiral side chain onto the peripheral moiety of phthalocyanine ring resulting in various tetra-and octa- β-substituted phthalocyanines [5a,b]. However, non-peripherally α-substituted phthalocyanines by chiral side chain have been rarely reported so far, to the best of our knowledge.
In the present paper, chiral menthol substituents are incorporated onto the non-peripheral moiety of phthalocyanine ring, resulting in a pair of "pinwheel-like" metal free phthalocyanines, (D)-and (L)-1, 8, 15, 22-tetrakis (2-isopropyl-5-methylcyclohexoxyl) phthalocyanine (1, Scheme 1). The newly prepared phthalocyanine has been characterized by a series of spectroscopic technique. In addition, their electrochemical properties have also been studied by cyclic voltammetry measurement.

|
Download:
|
| Scheme 1. Synthesis of metal-free 1, 8, 15, 22-tetrakis (2-isopropyl-5-methylcyclohexoxyl) phthalocyanine enantiomers (D)-1 and (L)-1. | |
Phthalocyanine compounds could be prepared through several possible synthetic pathways. The most commonly used method is the so-called tetra-cyclization of the corresponding phthalonitrile precursor. Herein, the chiral precursor 3-(2-isopropyl-5-methylcyclohexoxyl)-1, 2-dicyanobenzene (2) for the synthesis of the phthalocyanines was synthesized by a simple one-step procedure starting from 3-nitrophthalonitrile and (D)-or (L)-menthol in good yield. In a tetra-cyclization method, both (D)-and (L)-enantiomers of the target metal-free phthalocyanine 1 were isolated in relatively good yield from the cyclic tetramerization of (D)-and (L)-2, respectively, in refluxing n-pentanol in the presence of lithium followed by treatment with acetic acid (Scheme 1). The newly prepared chiral phthalocyanines were soluble in common organic solvents such as CHCl3, CH2Cl2, and toluene, and could therefore be purified readily by column chromatography. Interestingly, cyclisation of 3-substituted phthalonitriles usually gives a mixture of four constitutional isomers of tetra-substituted phthalocyanines, however, only C4 tetra-α-substituted phthalocyanine 1 has been predominantly generated and isolated in the present case, possibly owing to the steric hindrance of the bulky menthol groups results in other isomers unstable [6].
Satisfactory elemental analysis results were obtainedforboth the newly metal free (D)-and (L)-phthalocyanines. The enantiomers were further characterized by MALDI-TOF mass, 1H NMR spectroscopic and IR spectroscopic. TheMALDI-TOFmass spectra of 1 clearly showed intense signals for molecular ion[M]+. The isotopic pattern closely resembled the simulated one given in Fig. S1(Supporting information).
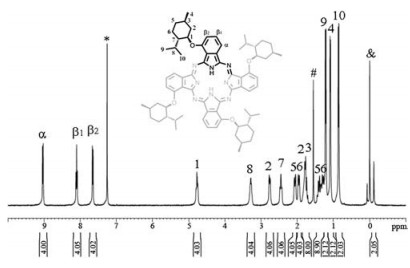
As would normally be expected, the two enantiomers of metal free phthalocyanine 1 have identical NMR, electronic absorption, and IR spectroscopic spectra. Only the data for the (L)-1 are, therefore, discussed. Fig. 1 shows the 1H NMR of the 1 recorded in CDCl3. With the help of the 1H-1H COSY spectrum, Fig. S2 (Supporting information), all the signals could be unambiguously assigned with the result detailed in Table S1(Supporting information). The 1H NMR spectrum of 1 exhibited one doublet at δ 9.03 due to the Pc α protons and one triplet at δ 8.10 and one doublet at δ 7.65 due to the Pc β1 and β2 protons, respectively, which are well defined and correlated with each other, revealing the C4 symmetry and the tetra-α-substituted phthalocyanine nature. Also, on the basis of the analysis over the 1H-1H COSY spectrum of 1, the aliphatic proton signals of menthol substituents, can be unambiguously assigned. As can be seen in Fig. 1 and Table S1, the signal appearing at δ 4.77 is assigned to the methenyl protons H1 close to the oxygen atoms, which is correlated with the signals at δ 2.76, 1.72-1.79, and 2.45 due to the methylene protons H2 and methenyl protons H7, respectively. Signals for the isopropyl protons H8-H10 exhibit one multiplets at δ 3.28 and two doublets at δ 1.21 and 0.86. The methyl protons H4 signal was observed at δ 1.08, correlated with signal for the methenyl protons H3 at δ 1.72-1.79, while the methylene protons H5 and H6 give several multiplets at δ 2.06, 1.95, and 1.23-1.50.In addition, the singlet at δ-0.12 was assigned to the inner isoindole protons.

|
Download:
|
| Fig. 1. 1H NMR spectrum for 1 in CDCl3. The signals due to residue CHCl3, H2O, and SiMe4 are denoted as*, #, and &, respectively. | |
It has been proved that vibrational spectroscopy is a versatile technique for studying the intrinsic properties of phthalocyanine compounds. In the IR spectrum of 1(Fig. S3 in Supporting information), the absorption bands contributed from the central aromatic Pc macrocycle was observed, including the wagging and torsion vibrations of C-H groups at 672-753 cm-1, isoindole ring stretching vibrations 1070-1420 cm-1, and the C=N aza group stretching vibrations at 1584-1596 cm-1 [7]. The absorptions observed at ca. 2960, 2928, and 2866 cm-1 are assigned to the asymmetric and symmetric C-H stretching vibrations, while the signals at 1275 and 1095 cm-1 are due to the asymmetric and symmetric C-O-C stretching vibrations of the menthol groups [8]. In addition, a weak absorption at 3294 cm-1 is obviously due to the asymmetrical N-H stretching vibration [8].
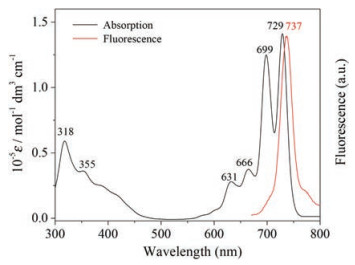
The electronic absorption spectrum for the metal free phthalocyanine 1 was recorded in CH2Cl2. As can be seen in Fig. 2, compound 1 shows typical features of the electronic absorption spectra for metal free phthalocyanines with splited Q bands at 729 and 699nm accompanied by two weak vibronic shoulder at 666 and 631nm and medium intensity Soret bands appearing at 355 and 318nm. Note that the Q bands of 1 are significantly red-shifted compared to the unsubstituted phthalocyanine and the β-substituted phthalocyanine by alcoxyl chain, owing to the α-substituted nature [8, 9]. The weak broad band appearing at around 450nm is due to the n-π*transitions arising from the oxygen lone pairs of electrons, which is common for alkoxy and phenoxy substituted phthalocyanines [8]. Unfortunately, like other phthalocyanines containing chiral carbons in the side chain [5a,b], neither the (D)-1 nor (L)-1 enantiomer displays a CD signal in the Soret and Q absorption region of the phthalocyanine ligand, (Fig. S4 in Supporting information), indicating the lack of effective chiral information transfer from the chiral menthol side chains to the phthalocyanine chromophore at the molecular level. In addition, it can be seen that the metal free phthalocyanine 1 exhibits intense mirror-imaged emissions at 737nm upon excitation at 630nm with the fluorescence quantum yield of 21% in CH2Cl2.

|
Download:
|
| Fig. 2. Electronic absorption (black line) and fluorescence spectra (red line) of 1 in CH2Cl2. | |
The electrochemical behaviour of 1 was investigated by cyclic voltammetry (CV) in CH2Cl2 containing 0.1mol/L [Bu4N][PF6]. Fig. S5(Supporting information) shows the CV of 1 and the halfwave redox potential data vs. SCE are summarized in Table 1. As can be seen, within the electrochemical window of CH2Cl2, compound 1 undergo two quasi-reversible one-electron reductions and two quasi-reversible one-electron oxidations since the separation between the reduction and oxidation peak potentials for each process is 55-100mV. The CV of compound 2 was also recorded in CH2Cl2 under the same condition (Fig. S6 in Supporting information), which shows neither reduction peak nor oxidation peak, indicating the redox waves of 1 shown in Fig. S5(Supporting information) are all assigned to the phthalocyanine ring. In comparison with unsubstituted phthalocyanine, the first oxidation and first reduction potentials gradually shift to the negative direction due to the electron-donoring nature of the menthol moiety, leading to the difference between the first oxidation and first reduction (△Eθ1/2) of 1.59 V for 1, smaller than that for unsubstituted phthalocyanine [9]. This result is also in agreement with the red-shift of lowest energy absorption in the Q bands from 700nm for unsubstituted phthalocyanine to 729nm for 1.
|
|
Table 1 Half-wave redox potentials of 1 recorded in CH2Cl2 containing 0.1mol/L [Bu4N]-[ClO4]at a scan rate of 50mV/s.a |

{kind=link}
{kind=link}
{kind=link}
In summary, we have prepared a pair of "pinwheel-like" metal free phthalocyanines with four non-peripherally attached chiral menthol units, which have been characterized by a series of spectroscopic technique. The steric hindrance of the bulky menthol groups resulted in the C4 tetra-α-substituted phthalocyanine with defined molecular structure is the main phthalocyanine product, which has been purified readily by column chromatography and revealed by NMR spectroscopy. This work is helpful for the design and synthesis of new non-peripherally α-substituted phthalocyanines by chiral side chain with various potential applications.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 41771342 and 21607096), and Funds of Shandong "Double Tops" Program (No. SYL2017XTTD15).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.06.002.
| [1] |
(a) A. Guijarro, M. Yus, The Origin of Chirality in the Molecules of Life, RSC Publishing, Cambridge, 2009; (b)U. Meierhenrich, Amino Acids and the Asymmetry of Life, Springer-Verlag, Berlin, Heidelberg, 2008; (c)W. J. Lough, I. W. Wainer, Chirality in Natural and Applied Science, Blackwell Science, Oxford, 2002. |
| [2] |
(a) M. Wang, G. L. Silva, B. A. Armitage, J. Am. Chem. Soc. 122 (2000) 9977-9986; (b)R. F. Pasternack, A. Giannetto, P. Pagano, E. J. Gibbs, J. Am. Chem. Soc. 113 (1991) 7799-7800; (c)K. C. Hannah, B. A. Armitage, Acc. Chem. Res. 37 (2004) 845-853; (d)X. Chen, M. Liu, J. Inorg. Biochem. 94 (2003) 106-113; (e)J. D. Watson, F. H. C. Crick, Nature 171 (1953) 737-738. |
| [3] |
(a) H. Ogoshi, T. Mizutani, Acc. Chem. Res. 31 (1998) 81-89; (b)G. Simonneaux, P. L. Maux, Coord. Chem. Rev. 228 (2002) 43-60; (c)G. A. Hembury, V. V. Borovkov, Y. Inoue, Chem. Rev. 108 (2008) 1-73; (d)Y. Furusho, T. Kimura, Y. Mizuno, T. Aida, J. Am. Chem. Soc. 119 (1997) 5267-5268; (e)Y. Mizuno, T. Aida, K. Yamaguchi, J. Am. Chem. Soc. 122 (2000) 5278-5285; (f)A. Mammana, A. D'Urso, R. Lauceri, R. Purrello, J. Am. Chem. Soc. 129 (2007) 8062-8063; (g)F. Helmich, C. C. Lee, A. P. H. J. Schenning, E. W. Meijer, J. Am. Chem. Soc. 132 (2010) 16753-16755. |
| [4] |
(a) B. Meunier, Chem. Rev. 92 (1992) 1411-1456; (b)J. P. Collman, X. Zhang, V. J. Lee, E. S. Uffelman, J. I. Brauman, Science 261 (1993) 1404-1411; (c)J. P. Collman, R. Boulatov, C. J. Sunderland, L. Fu, Chem. Rev. 104 (2004) 561-588; (d)H. Lu, X. P. Zhang, Chem. Soc. Rev. 40 (2011) 1899-1909. |
| [5] |
(a) N. Kobayashi, Coord. Chem. Rev. 219-221 (2001) 99-123; (b)H. Lu, N. Kobayashi, Chem. Rev. 116 (2016) 6184-6261; (c)K. Wang, D. Qi, H. Wang, et al., Chem. Eur. J. 18 (2012) 15948-15952; (d)H. Zhou, K. Wang, D. Qi, J. Jiang, Dalton Trans. 43 (2014) 1699-1705; (e)W. Lv, P. Zhu, Y. Bian, et al., Inorg. Chem. 49 (2010) 6628-6635; (f)K. Wang, S. Zeng, H. Wang, J. Dou, J. Jiang, Inorg, Chem. Front. 1 (2014) 167-171. |
| [6] |
(a) Y. Bian, R. Wang, J. Jiang, et al., Chem. Commun. (2003) 1194-1195; (b)Y. Bian, L. Li, J. Dou, et al., Inorg. Chem. 43 (2004) 7539-7544. |
| [7] |
R. Jiang, M. Bao, D. Rintoul, Arnold, Coord. Chem. Rev. 250 (2006) 424-448. DOI:10.1016/j.ccr.2005.09.009 |
| [8] |
(a) H. Pan, C. Chen, K. Wang, W. Li, J. Jiang, Chem. Eur. J. 21 (2015) 3168-3173; (b)Y. Zhang, X. Zhang, Z. Liu, Y. Bian, J. Jiang, J. Phys. Chem. A 109 (2005) 6363-6370; (c)N. Sheng, R. Li, C. F. Choi, et al., Inorg. Chem. 45 (2006) 3794-3802; (d)Q. Liu, Q. Zhao, Y. Li, X. Wang, CrystEngComm 14 (2012) 1105-1110. |
| [9] |
R. Li, X. Zhang, P. Zhu, et al., Inorg. Chem. 45 (2006) 2327-2334. DOI:10.1021/ic051931k |