 2019, Vol. 30
2019, Vol. 30 


1, 2, 3-Thiadiazol-5-ylureas are a class of heterocyclic compounds containing both heteroatoms such as N, O, S and peptide bonds in their molecules. Therefore, most of them have biological activities and are used primarily as plant growth regulators in agriculture [1-4]. For example, as an important plant growth regulator, 1-phenyl-3-(1, 2, 3-thiadiazol-5-yl)urea (thiadiazuron) not only can be used as a defoliant in cotton [5, 6] but also has been proved to be very effective in stimulating kiwifruit [7, 8] and pear growth [9]. In addition, some of these compounds also have herbicidal activities [10], fungicidal activities [11] and anticancer activities [12].
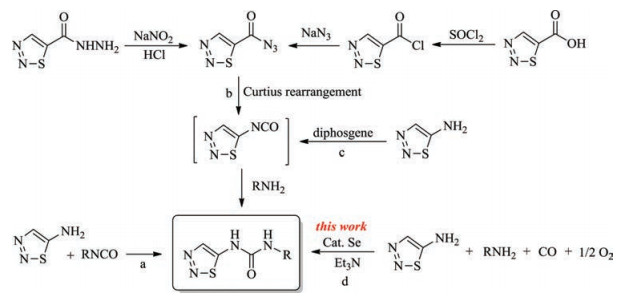
So far, the main approaches to these compounds reported in literatures include: 1) addition of 1, 2, 3-thiadiazol-5-amine to isocyanates (Scheme 1, route a) [7]; 2) addition of amines to 5- isocyanato-1, 2, 3-thiadiazole generated in situ via Curtius rearrangement of 1, 2, 3-thiadiazole-5-carbonyl azide which is obtained from 1, 2, 3-thiadiazole-5-carbohydrazide [13] or 1, 2, 3-thiadiazole- 5-carboxylic acid [8] by multi-step reactions (Scheme 1, route b); 3) addition of amines to 5-isocyanato-1, 2, 3-thiadiazole generated in situ by the reaction of 1, 2, 3-thiadiazol-5-amine with diphosgene (Scheme 1, route c) [14]. However, there are many drawbacks associated with these methods such as complex structure and inconvenient sources of the raw materials, tedious synthetic route, cumbersome operations, low atomic economy, high cost and generation of corrosive acid wastes.

|
Download:
|
| Scheme 1. Previous and present synthetic approaches to 1, 2, 3-thiadiazol-5-ylureas. | |
As the most important C1 building block, CO has been employed extensively for various carbonylation transformations in the presence of catalysts [15-20]. Over the past decades, cheap and readily available nonmetal selenium has been found and applied as an effective catalyst for the carbonylation reaction with CO to prepare many valuable carbonyl-containing compounds such as ureas [21-23], carbamates [21, 24, 25], thiocarbamates [21, 26] and carbonates [21, 27] with high atom economy in one-pot manner. Recently, Se-carbonylation reaction has also been developed for the preparation of 1, 3-selenazolidin-2-ones [28]. However, no example has so far been found in literatures on the synthesis of 1, 2, 3-thiadiazol-5-ylureas with Se/CO catalytic system. Herein, we wish to report a facile and economical approach to these target compounds via one-pot selenium-catalyzed oxidative carbonylation of 1, 2, 3-thiadiazol-5-amine with various amines in the presence of CO and O2 (Scheme 1, route d).
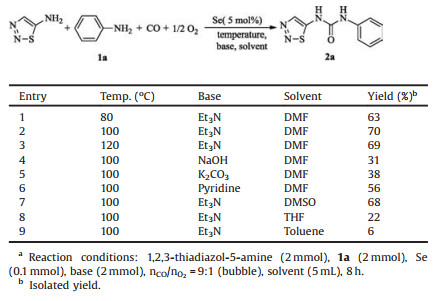
Initially, the oxidative carbonylation of 1, 2, 3-thiadiazol-5- amine with aniline (1a) was chosen as a model reaction to optimize the reaction conditions (Table 1). For safety reason, the ratio of CO to O2 was designated as 9:1 during the seleniumcatalyzed oxidative carbonylation reaction. To our delight, the reaction could proceed smoothly at 80℃ and afforded the target product 1-phenyl-3-(1, 2, 3-thiadiazol-5-yl)urea (2a) in 63% yield (Table 1, entry 1) accompanied with a small amount of diphenylurea and di(1, 2, 3-thiadiazol-5-yl)urea, suggesting that accompanying by the cross carbonylation reaction there existed the competitive carbonylation of 1, 2, 3-thiadiazol-5-amine and aniline themselves. When the temperature was raised to 100℃, higher product yield could be obtained (Table 1, entry 2). Further increase of the temperature to 120℃ failed to gain the beneficial result (Table 1, entry 3). Generally, a proper alkaline condition is believed to be necessary for the generation of the active specie carbonyl selenide (COSe) in the selenium-catalyzed carbonylation reactions [21, 29]. Thus, the common alkalis such as NaOH, K2CO3, Pyridine and Et3N were screened (Table 1, entries 2, 4–6) and the results indicated that the organic alkalis (Table 1, entries 2, 6) were more effective than those inorganic ones (Table 1, entries 4, 5). Among them, Et3N worked best. Finally, the effect of solvents on the carbonylation reaction was also studied (Table 1, entries 2, 7– 9). According to the results, strong polar solvents worked better (Table 1, entries 2, 7) than the weak ones (Table 1, entries 8, 9). So, DMF was chosen ultimately as the solvent in this carbonylation system for convenience purpose.
|
|
Table 1 Screening of optimized conditionsa. |
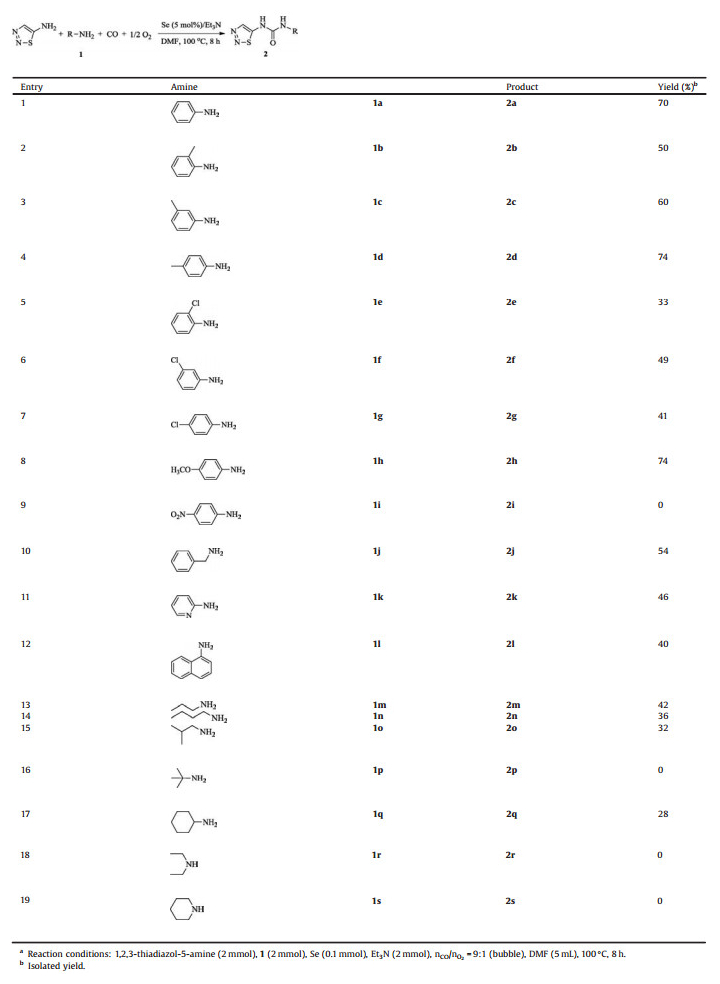
With the optimized reaction conditions in hand, we next investigated the scope and efficiency of the selenium-catalyzed oxidative carbonylation of 1, 2, 3-thiadiazol-5-amine with a series of amines (Table 2). Experimental details and characterization data of the target compounds are presented in Supporting information. The results indicated that most of the amines could tolerate the carbonylation reaction, affording the corresponding 1, 2, 3-thiadiazol-5-ylureas in moderate to good yields. Firstly, aromatic amines were applied in the carbonylation reaction and the results revealed that the reaction was very sensitive to both electronic effect and steric effect of the aromatic amines. For example, the anilines bearing electron-donating group (Table 2, entries 2–4 and 8) such as 4-methylbenzenamine and 4-methoxylbenzenamine (Table 2, entries 4 and 8) were more effective than those with electronwithdrawing group (Table 2, entries 5–7 and 9) such as 4- cholrobenzenamine and 4-nitrobenzenamine (Table 2, entries 7 and 9), resultingin the higher product yields of the former than the latter. As for steric effect, ortho-substituted anilines (Table 2, entries 2, 5) were less effective than those meta-substituted or para-substituted ones (Table 2, entries 3, 4, 6, 7), resulting in the lower product yields of the former than the latter. As the examples of heterocyclic amines and polycyclic amines, pyridin-2-amine and naphthalen-1-amine were also applied in this selenium-catalyzed system. The results indicated that both of them could tolerate the carbonylation reaction, affording the corresponding product in yield of 46% (Table 2, entry 11) and 40% (Table 2, entry 12), respectively. Next, the substrate scope of the carbonylation reaction was extended to aliphatic amines. On the whole, aliphatic amines worked less efficiently (Table 2, entries 13–19) than those aromatic ones most probably due to their weak reactivities. Steric effect seemed to affect the carbonylation reaction significantly. Less hindered aliphatic amines such as propan-1-amine and butan-1-amine worked more efficiently (Table 2, entries 13, 14) than those more hindered ones such as 2-methylpropan-1-amine and cyclohexanamine (Table 2, entries 15, 17). The greater the steric hindrance of the aliphatic amines is, the lower their reactivity becomes. Thus, it was understandable that no desired products were obtained when the carbonylation reaction proceeded with the highly hindered 2-methylpropan-2-amine (Table 2, entry 16) as well as two secondary amine examples of diethylamine and piperidine (Table 2, entries 17, 18).
|
|
Table 2 Amine scope of the selenium-catalyzed carbonylation reaction.a |
{kind=link}
Compared with other catalytic systems, one remarkable characteristic of this selenium-catalyzed carbonylation system is that it has the function of phase-transfer catalysis. In particular, selenium powder is insoluble in solvent prior to the carbonylation reaction; then it can react with CO in proper alkaline condition to form soluble carbonyl selenide (SeCO) in situ, which initiate the subsequent homogeneous carbonyaltion reaction to form the target product during the reactionprocess; upon the completion of the reaction, insoluble selenium power can be precipitated from the reaction mixture after sufficient oxidation and can be easily recovered by suction filtration. Thus, the reaction system can change reciprocally between heterogeneous and homogeneous systems during the whole reaction process, which can not only realize the efficient catalytic reaction, but also facilitate the separation and recovery of the catalyst selenium. The reusability of the recovered selenium was tested via the model carbonylation reaction of 1, 2, 3-thiadiazol-5-amine with aniline. The yield of 1- phenyl-3-(1, 2, 3-thiadiazol-5-yl)urea (2a) only dropped from 70% to 65% after four cycles, which proved the catalytic performance of the recovered selenium was almost the same as that of the fresh selenium considering its small loss in each recovery step.
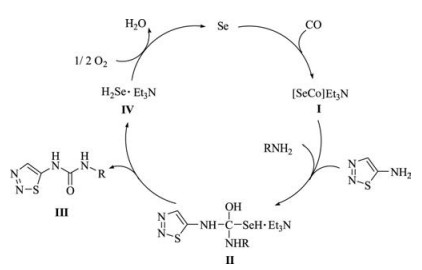
A plausible reaction mechanism of selenium-catalyzed oxidative carbonylation to 1, 2, 3-thiadiazol-5-ylureas is illustrated in Scheme 2. Firstly, the carbonylation reaction is initiated by the active specie carbonyl selenide (Ⅰ), which is generated in situ via the reaction of Se with CO in the presence of triethylamine [21]. Next, nucleophilic attack of 1, 2, 3-thiadiazol-5-amine and the amine on Ⅰ forms the intermediate Ⅱ, which then undergoes elimination to afford the target product Ⅲ, accompanied by the formation of hydrogen selenide (Ⅳ). Specie Ⅳ is then oxidized to Se by oxygen for the ensuing catalytic cycle.

|
Download:
|
| Scheme 2. Proposed reaction pathway to 1,2,3-thiadiazol-5-ylureas. | |
{kind=link}
In summary, a facile one-pot, economical approach to 1, 2, 3- thiadiazol-5-ylureas has been developed. With cheap and easily available 1, 2, 3-thiadiazol-5-amine and amines as raw materials, cheap and reusable selenium as the catalyst, carbon monoxide as the carbonylation reagent and oxygen as the oxidizing agent, the selenium-catalyzed oxidative carbonylation reaction of 1, 2, 3- thiadiazol-5-amine can proceed smoothly with a series of amines in one-pot manner in the presence of triethylamine, affording the target products 1, 2, 3-thiadiazol-5-ylureas mostly in moderate to good yields. The features associated with this approach such as low cost, high atomic economy, one-pot approach, simple operations and no emission of corrosive wastes should make it very promising towards this class compounds.
AcknowledgmentsWe are grateful for the financial support from the National Natural Science Foundation of China (No. 21772033) and Program for Innovative Research Team in Science and Technology in University of Henan Province (No. 15IRTSTHN003).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j. cclet.2018.07.017.
| [1] |
A. Kumari, P. Baskaran, L. Plackova, et al., J. Plant Physiol. 223 (2018) 65-71. DOI:10.1016/j.jplph.2018.01.005 |
| [2] |
S.K. Dwivedi, A. Arora, V.P. Singh, et al., Photosynthetica 56 (2018) 478-486. DOI:10.1007/s11099-017-0695-2 |
| [3] |
C.S. Schmidt, L. Mrnka, T. Frantik, et al., Plant Physiol. Biochem. 120 (2017) 120-131. DOI:10.1016/j.plaphy.2017.09.016 |
| [4] |
M.C. Mok, D.W.S. Mok, D.J. Armstrong, et al., Phytochemistry 21 (1982) 1509-1511. DOI:10.1016/S0031-9422(82)85007-3 |
| [5] |
Y.S. Yang, Y. Zhang, M.G. Dong, et al., J. Hazard. Mater. 335 (2017) 92-99. DOI:10.1016/j.jhazmat.2017.04.013 |
| [6] |
K. Grossmann, Plant Physiol. 95 (1991) 234-237. DOI:10.1104/pp.95.1.234 |
| [7] |
Z.W. Zhang, H.H. Yang, Z.H. Gao, et al., J. Agric. Food Chem. 65 (2017) 11273-11279. DOI:10.1021/acs.jafc.7b03522 |
| [8] |
A. Abad, C. Agullo, A.C. Cunat, et al., J. Agric. Food Chem. 52 (2004) 4675-4683. DOI:10.1021/jf049921+ |
| [9] |
M.S. Pasa, A.F. Brighenti, C.P.D. Silva, et al., An. Acad. Bras. Cienc. 89 (2017) 3103-3110. DOI:10.1590/0001-3765201720170644 |
| [10] |
R. Mirzajani, Z. Ramezani, F. Kardani, Microchem. J. 130 (2017) 93-101. |
| [11] |
A.D. Pavlista, Acta Hortic. 619 (2003) 145-152. |
| [12] |
G. Enkhtaivan, D.H. Kim, M. Pandurangan, J. Photochem. Photobiol. B Biol. 173 (2017) 493-498. DOI:10.1016/j.jphotobiol.2017.06.032 |
| [13] |
T.A. Kalinina, Y.S. Shakhmina, T.V. Glukhareva, et al., Chem. Heterocycl. Com+. 50 (2014) 1039-1046. DOI:10.1007/s10593-014-1561-9 |
| [14] |
J. Nisler, M. Zatloukal, L. Spichal, et al., Patent, WO 2016037595 A1, 2016.
|
| [15] |
X.F. Wu, X.J. Fang, L.P. Wu, et al., Acc. Chem. Res. 47 (2014) 1041-1053. DOI:10.1021/ar400222k |
| [16] |
Q. Liu, H. Zhang, A.W. Lei, Angew. Chem. Int. Ed. 50 (2011) 10788-10799. DOI:10.1002/anie.v50.46 |
| [17] |
A. Brennfuhrer, H. Neumann, M. Beller, Angew. Chem. Int. Ed. 48 (2009) 4114-4133. DOI:10.1002/anie.v48:23 |
| [18] |
X.F. Wu, H. Neumann, M. Beller, ChemSusChem 6 (2013) 229-241. DOI:10.1002/cssc.v6.2 |
| [19] |
K. Shi, S.Y. Huang, Z.Y. Zhang, et al., Chin. Chem. Lett. 28 (2017) 70-74. DOI:10.1016/j.cclet.2016.06.005 |
| [20] |
H.T. Zhang, L.J. Gu, X.Z. Huang, et al., Chin. Chem. Lett. 27 (2016) 256-260. DOI:10.1016/j.cclet.2015.10.012 |
| [21] |
N. Sonoda, Pure Appl. Chem. 65 (1993) 699-706. DOI:10.1351/pac199365040699 |
| [22] |
T. Mizuno, T. Nakai, M. Mihara, Synthesis (2010) 4251-4255. |
| [23] |
X.P. Zhang, Z.J. Tang, X.L. Niu, et al., Tetrahedron Lett. 57 (2016) 5266-5270. DOI:10.1016/j.tetlet.2016.10.046 |
| [24] |
X.P. Zhang, H.Z. Jing, J. Mol. Catal. A Chem. 302 (2009) 137-141. DOI:10.1016/j.molcata.2008.12.012 |
| [25] |
X.P. Zhang, H.Z. Jing, W.Y. Peng, et al., Chin. J. Org. Chem. 37 (2017) 411-417. DOI:10.6023/cjoc201609013 |
| [26] |
X.P. Zhang, H.Z. Jing, S.W. Lu, Synlett (2005) 1535-1538. |
| [27] |
T. Mizuno, T. Nakai, M. Mihara, Heteroat. Chem. 21 (2010) 541-545. DOI:10.1002/hc.v21:7 |
| [28] |
H. Wang, J. Ying, M. Lai, et al., Adv. Synth. Catal. 360 (2018) 1693-1703. DOI:10.1002/adsc.v360.8 |
| [29] |
N. Sonoda, T. Yasuhara, K. Kondo, et al., J. Am. Chem. Soc. 93 (1971) 6344. DOI:10.1021/ja00752a099 |