 2019, Vol. 30
2019, Vol. 30 


b Key Laboratory of Metabolomics at Shenzhen, Shenzhen 518055, China;
c School of Medicine, Tsinghua University, Beijing 100084, China
Breast cancer as a solid tumor is among the three of most common cancers worldwide and the most common malignancy in women [1]. Currently, multicellular tumor spheroids (MCTS) are the most widely used three-dimensional (3D) tumor model in preclinical studies, which recapitulate the biological and physiological properties of solid tumors [2]. To the best of our knowledge, tumor microenvironment plays a crucial role in tumor initiation, progression, and metastasis [3]. Among which, macrophages and extracellular matrix (ECM) have been reported as two key components in the tumor microenvironment regulating tumor cell behaviors. Many researches have showed that tumorassociated macrophages (TAMs) are a macrophage subset that directly contribute to tumor invasion [4, 5], tumor angiogenesis [6], and ECM remodeling [7]. In addition, clinical studies have confirmed the correlation of high infiltration of TAMs in tumor stroma with poor prognosis in case of breast cancer [8]. ECM, a complex network of macromolecules, is commonly deregulated and becomes disorganized in diseases such as cancer [9]. ECM stiffness as an important biomechanical parameter also affects endothelial-mesenchymal transition and tumor cell migration, proliferation, apoptosis, and so on [10, 11]. A recent study on the testing human breast biopsies showed that malignant tissues had a broad stiffness distribution because of tissue heterogeneity, while the cancer cells located in the microenvironment with a prominent low-stiffness peak [12]. Moreover, tumor development to the late, invasive stage aggravated the soft peak and further broadened the stiffness distribution. Therefore, ECM stiffness may provide a mechanobiological indicator in the clinical diagnostics.
Due to the important roles of macrophages and ECM stiffness in cancer development, conventional in vitro models like transwell assays [13, 14] have been developed to study their impacts on tumor cell migration and invasion. However, these models failed to reconstruct the appropriate 3D tumor microenvironment due to lack of cell-cell and cell-matrix interactions [15]. Referring toMCTS formation, the common methods including liquid overlay [16], hang drop [17], and spinner flask [18] encountered weakness such as difficulty in the control of uniform spheroid size, timeconsuming process, unavailable for real-time and in situ detection [19]. In recent years, microfluidics has been widely employed in cellular biology research due to its inherent advantages such as flexible cell manipulation, long-term cell culture and accessibility to combine with many analytical techniques [20-22]. A previous study showed that the effects of macrophages on cancer cells migration through ECM on a microfluidic chip, but cancer cells were only cultured in 3D matrix rather than the spheroids mode which is closestto in vivo solid tumors[23].Recently, a microfluidic system integrating tumor cell aggregates and macrophages in collagen matrix was reported to reveal different subtypes of macrophages-tumor cell interactions, but aggregates were formed in the laser-ablated microwells and then transferred to the microchannels, which made the manipulation more complex [24]. Although tumor spheroids could be formed in situ in the microchambers, they varied greatly in size [25]. Additionally, the co-effects of ECM and TAMs on tumor cell behaviors have not been studied in detail. Therefore, there is an urgent need to develop an appropriate model to reveal the relationships among macrophages, ECM and tumor cells.
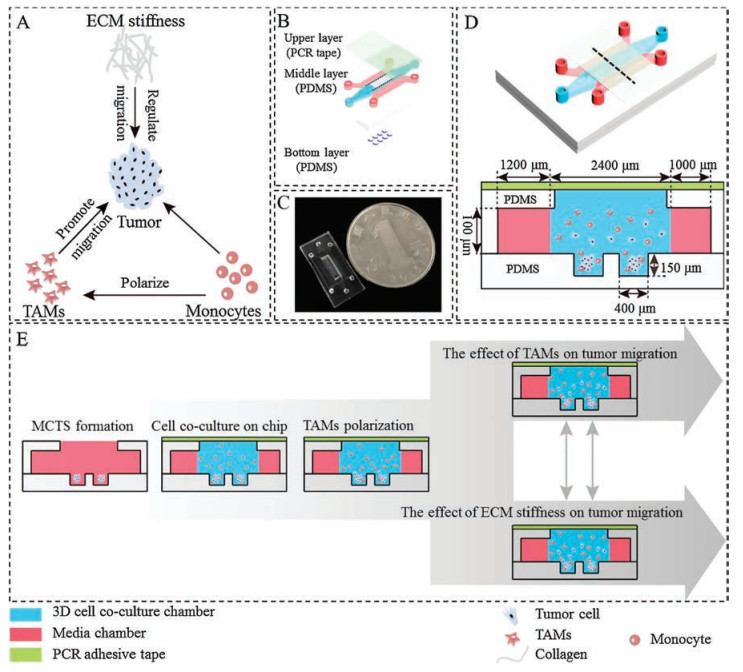
Herein, in order to investigate the effects of TAMs and ECM stiffness on tumor cell migration (Fig. 1A), a three-layer microfluidic device containing two PDMS layers and a PCR adhesive tape was designed and fabricated (Figs. 1B–D). The bottom PDMS layer contained microwells for MCTS formation. The middle layer had threechambers separated by trapezoidal PDMS columnsfor 3D cell co-culture and nutrient supply. In order to help MCTS formation easily in the bottommicrowells, a rectangularopen areawas cuton the middle chamber of the middle PDMS layer which was aligned with the bottom microwells. The bottom and middle PDMS layers were finally irreversibly bonded after oxygen plasma treatment. The microwells with 400 μm in diameter and 150 μm in depth were designed to accommodate a sufficient number of cells and prevent MCTS being washed out from the microwells when refreshing medium. After formation of MCTS in the microwells or co-culturewith monocytes, the rectangularopen areaneeded tobe sealed with a PDMS substrate or other materials to form a closed chamber for further study. Inspired by a recent study [26], a PCR adhesive tape was employed for reversible bonding without accessing specific equipment or protocols. Due to the reversible bonding, cells could be conveniently collected for further off-chip analysis. The workflow of 3D cell co-culture in microfluidics was presented in Fig. 1E.

|
Download:
|
| Fig. 1. Schematic illustrations of the 3D co-culture microfluidic device. (A) Intricate interactions of cellular and non-cellular components of the tumor microenvironment. (B) The device consists of two PDMS layers and a PCR adhesive tape. (C) A photo of microfluidic device. (D) The overall schematic presentation and the side view of the microfluidic device. (E) Workflow of 3D co-culture microfluidic model construction and tumor cell migration study. | |
{kind=link}
The tumor microenvironment has been recognized as a key regulator of carcinogenesis, including ECM and numerous types of stromal cells like macrophages. To probe the effects of macrophages and ECM stiffness on breast tumor cell migration, low invasive T47D and high invasive MDA-MB-231 breast carcinoma cells were selected to form MCTS in the microwells. Prior to cell seeding, 1% (w/v) polyvinyl alcohol (PVA) solution was used to modify PDMS to reduce cell adhesion and promote MCTS generation [27]. Monodisperse breast tumor spheroids were formed in the microwells during the first 24 h (Fig. S1 in Supporting information). The use of open system allowed easier injection of PVA solution and cell suspension to the microwells. As shown in Fig. 2A, T47D and MDA-MB-231 cells could form spheroids in the 1.5 mg/mL of collagen with uniform diameter of greater than 200 μm, while the diameter of MDA-MB-231 spheroids was a little larger than that of T47D spheroids. Compared to the irreversibly bonded device, MCTS could not retain in the microwells and aggregated when collagen was added into the chamber (Fig. S2 in Supporting information). In order to investigate the cell viability of MCTS over long-term cultivation, T47D MCTS were imaged every day. At day 7, more than 95% of cells were alive (Fig. S3 in Supporting information). The co-culture of T47D or MDA-MB-231 MCTS with U937 cells in 3D collagen matrix was also realized on the reversibly bonded microfluidic device (Fig. 2B). The results demonstrated that our established platform had obvious advantages in the formation of uniformly sized spheroids and the coculture of cells in 3D collagen matrix.

|
Download:
|
| Fig. 2. (A) Optical image of T47D and MDA-MB-231 MCTS generated in microwell arrays with uniform size in 1.5 mg/mL collagen for 3 h. (B) A image showed the distribution of two cell types (tumor cells and monocytes) in 1.5 mg/mL collagen in co-culture condition for 3 h. (C, D) CD68 and CD163 mRNA expression of U937 cells at 48 h by RT-PCR analysis when U937 cells were co-cultured without or with T47D MCTS. T47D MCTS in the presence of 5 ng/mL of M-CSF, MDA-MB-231 MCTS in 1.5 mg/mL of collagen. RT-PCR representative images (C) and quantification (D) were shown (mean ± SEM, n = 3 independent experiments; ***P < 0.001, ns: no sense). | |
{kind=link}
Monocytes are direct precursors of macrophages [28]. After their recruitment into the tumor tissue, monocytes can be polarized into TAMs by the regulation of cytokines, chemokines and growth factors which are derived from cancerous and noncancerous cells in the tumor microenvironment [29]. To explore whether breast cancer cells with distinct invasiveness have different abilities to polarize monocytes into TAMs, T47D and MDA-MB-231 MCTS were co-cultured with U937 cells separately in 1.5 mg/mL of collagen in the microfluidic device. To our knowledge, CD68 as a total macrophage marker and CD163 as a M2 macrophage marker were used to identify TAMs [30]. As shown in Figs. S4 A and B in Supporting information, U937 cells cocultured with T47D MCTS for 48 h remained round and unpolarized without the expression of macrophage marker CD68 protein. However, when U937 cells were co-cultured with MDA-MB-231 MCTS, they obtained TAM-like phenotype and polarized, and with the expression of CD68 protein. Macrophage colony-stimulating factor (M-CSF) is a cytokine that regulates the polarization and function of macrophages via its receptor M-CSFR [31]. Therefore, 5 ng/mL of M-CSF was added into the co-culture system of T47D MCTS and U937 cells to investigate whether U937 cells could be polarized into TAMs. As expected, U937 cells obtained TAM-like phenotype and polarized with CD68 protein expression. Furthermore, U937 cells were collected from the microfluidic device for RT-PCR assay. As shown in Fig. 2C, when U937 cells were cocultured with T47D MCTS, the expression of CD68 mRNA and CD163 mRNA were not detected. However, when U937 cells were co-cultured with high invasive MDA-MB-231 MCTS, the expression of CD68 mRNA and CD163 mRNA were both significantly upregulated. Meanwhile, T47D MCTS with the presence of M-CSF remarkably up-regulated the expression of CD68 and CD163 mRNA in U937 cells. This is in accordance with a recent study that M-CSFR signaling shaped the phenotype of M2-like TAM [32]. The corresponding quantification results were shown in Fig. 2D. The results suggested that high invasive MDA-MB-231 cells rather than low invasive T47D cells could polarize monocytes into TAMs. U937 cells co-cultured with T47D MCTS could also be activated to TAMs by the stimulation of M-CSF.
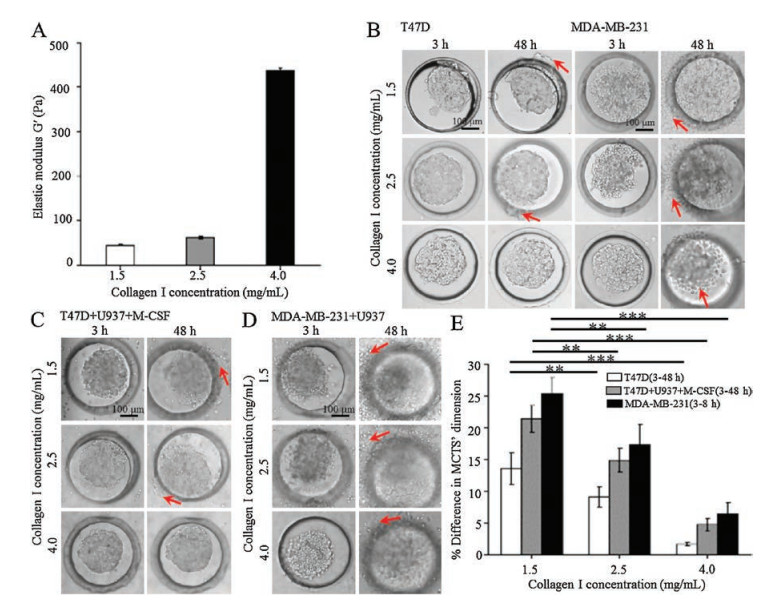
In order to investigate the effects ECM stiffness and TAMs on tumor cell migration, T47D and MDA-MB-231 MCTS were separately cultured with or without U937 cells in 3D collagen matrix with three different concentrations of 1.5 mg/mL, 2.5 mg/mL, and 4.0 mg/mL. The relationship between collagen concentration and stiffness was firstly investigated. As shown in Fig. 3A, the increased gel stiffness from 44.8 Pa to 436.1 Pa was generated by increasing the concentration of collagen gel from 1.5 mg/mL to 4.0 mg/mL. When spheroids were cultured in 3D collagen gel with different stiffness from 3 h to 48 h, T47D MCTS and MDA-MB-231 MCTS exhibited distinct migration abilities. As shown in Fig. 3B, referring to T47D MCTS, a poor migration phenomenon was observed with the decreasing of the collagen concentration between 3 h and 48 h. Only a small amount of T47D cells migrated in 1.5 mg/mL and 2.5 mg/mL of collagen gel. However, in terms of MDA-MB-231 MCTS, the cells had an obvious migrationphenomenon in three different concentrations of collagen. A large amount of MDA-MB-231 cells migrated out of the microwells in 1.5 mg/mL and 2.5 mg/mL of collagen gel, but the majority of cells in 4.0 mg/mL of collagen gel maintained at the original location. The results indicated that the migration of tumor cells was enhancedin softer 3D matrix. The synergetic effects of ECM stiffness and TAMs on tumorcell migrationwere furtherexplored.As shown in Fig. 3C and D, when T47D MCTS or MDA-MB-231 MCTS were co-cultured with U937 cells, the cell migration abilities were enhanced compared to that without U937 cells during the culture time from 3 h to 48 h. For example, T47D cells migration was enhanced in 1.5 mg/mL and 2.5 mg/mL of collagen gel with the presenceofU937cellsandM-CSFstimulation.However, thecellsstill exhibited no migration in 4.0 mg/mL of collagen gel even with the existenceofU937 cells.Incontrast, the migration ability of MDA-MB- 231 cells significantly increased in soft and rigid collagengel with the presence of TAMs. The results indicated that softer matrix and the presence of TAMs synergistically promoted tumor cell migration.The MCTS size (mean of major and minor axis length) was compared to the initial size to indicate the degree of the migration. The changes of spheroid size in Figs. S5A-C in Supporting information were quantified and the results were shown in Fig. 3E. The presence of U937 cells greatly increased MDA-MB-231 cells migration ability which could not be quantified, so that the result was not displayed. From the quantification results, we found that with the increasing of collagen concentration, the migration abilities of low invasive T47D cellsandhigh invasive MDA-MB-231 cellswith orwithout U937 cells all reduced. The reason might be contributed to the smaller pore size in the denser and stiffer 3D matrices which might impose considerable restrictions on the movement of cells and hence hinder the migration of the cells [33-35]. Under the same collagen concentration, MDA-MB-231 MCTS exhibited the strongest migration ability. The results indicated that even with the existence of U937 cells and M-CSF stimulation, the migration ability of low invasive T47D cells was weaker than that of high invasive MDA-MB- 231 cells.

|
Download:
|
| Fig. 3. The effects of ECM stiffness and TAMs on tumor cell migration. (A) Global shear elastic modulus of collagen matrix of varying concentrations. (B) Optical images of T47D and MDA-MB-231 MCTS grown in collagen of different concentrations at 3 h and 48 h. (C) Optical images of T47D MCTS grown in collagen of different concentrations with U937 cells in the presence of 5 ng/mL of M-CSF at 3 h and 48 h. (D) Optical images of MDA-MB-231 MCTS grown in collagen of different concentrations with U937 cells at 3 h and 48 h. (E) Percentage (%) change in different tumor spheroids' dimensions (average of major and minor axis length) from 3 h to 48 h (T47D mono-culture and co-culture condition) or from 3 h to 8 h (MDA-MB-231) (mean ± SEM, n = 4 independent experiments; **P < 0.01, and ***P < 0.001). Migratory cells were pointed out by red arrows. | |
{kind=link}
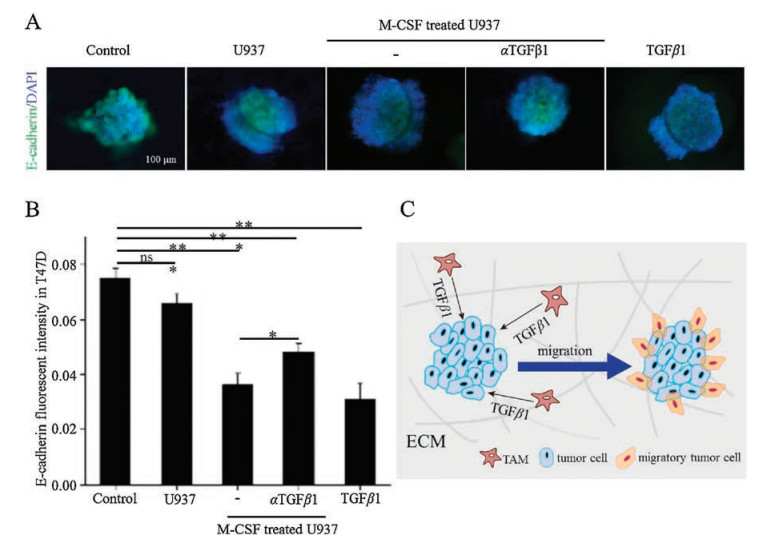
E-cadherin is a transmembrane protein that forms cell-cell junctions which are necessary for the adhesion and contractility of epithelial cells [36]. Previous studies found that down-regulation of E-cadherin expression in tumor cell may promote cell migration and invasion [37, 38]. To investigate the effect of ECM stiffness on the migration ability of tumor cells, E-cadherin protein in T47D tumor spheroids cultured in collagen of different concentrations was immunostained byanti-E-cadherin antibody. It was found that the expression of E-cadherin in T47D spheroids at 48h was downregulated when the spheroids were embedded in softer matrix, indicating that the migration of tumor cells was enhanced (Figs. S6A and B in Supporting information). To assess whether TAMs promoted tumor cell migration, the expression of E-cadherin in T47D MCTS was also evaluated. As shown in Figs. 4A and B, E– cadherin expression was not reduced prominently in the presence of U937 cells, whereas that in T47D MCTS co-cultured with M-CSF treated U937 cells was down-regulated markedly at 48h. The results indicated that the migration of T47D cells was enhanced by the presence of TAMs. TGFβ1 is considered to be a canonical cytokine correlated to tumor migration, metastasis and epithelial-mesenchymal transition (EMT) process [39], and a major secretory product of macrophages in the tumor microenvironment [40, 41]. The experiment whether TGFβ1 released from TAMs involved in promoting tumor cell migration was further performed. It was found that anti-TGFβ1 neutralizing antibody inhibited migration by up-regulating E-cadherin expression, whereas TGFβ1 promoted migration of T47D cells, suggesting that TGFβ1 is responsible for the ability of TAMs to promote tumor cell migration. Fig. 4C showed the proposed mechanism governing TAMs-promoted T47D cells migration via TGFβ1.

|
Download:
|
| Fig. 4. TAMs promoted the migration of T47D tumor cells via TGFβ1. (A) Fluorescent E-cadherin/DAPI staining at 48h inT47D MCTS in 1.5mg/mL collagenwithoutor with the co-culture of U937 cells, M-CSF-activated TAMs alone or with anti-TGFβ1 neutralizing antibody, 10ng/mL recombinant human TGFβ1. αTGFβ1 stands for anti-TGFβ1 neutralizing antibody. (B) E-cadherin fluorescent intensity in T47D MCTS (mean ±SEM, n=3 independent experiments; *P < 0.05, **P < 0.01, and ***P < 0.001, ns: no sense). (C) A diagram showing the possible mechanism governing TAMs-promoted T47D tumor cell migration via TGFβ1. | |
{kind=link}
In this study, we exploited a novel 3D co-culture model in a microfluidic device with reversible bonding to explore the effect of ECM stiffness and TAMs on tumor cell migration. This microfluidic system allowed rapid formation of breast MCTS and modulation of the tumor microenvironment by embedding MCTS in 3D collagen matrix with varied stiffness, in the presence or absence of TAMs. Using this established 3D breast cancer co-culture model, it was found that with increased ECM stiffness, breast MCTS reduced migration. On the other hand, M-CSF-activated TAMs rather than monocytes promoted the migration of breast tumor cells via the regulation of TGFβ1. Moreover, the softer matrix and TAMs synergistically contributed to the migration of tumor cells. In summary, this microfluidic platform provides a more biomimetic 3D model in vitro to replicate the tumor microenvironment in vivo using monocytes and MCTS co-culture system, which will help us better understand the tumor microenvironment and promote breast cancer therapeutic development.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21675096 and 21475073) and Youth Scientific Research Funds from Graduate School at Shenzhen, Tsinghua University (No. QN20160002).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.07.013.
| [1] |
N. Harbeck, M. Gnant, Lancet 389 (2017) 1134-1150. DOI:10.1016/S0140-6736(16)31891-8 |
| [2] |
S. Nath, G.R. Devi, Pharmacol. Ther. 163 (2016) 94-108. DOI:10.1016/j.pharmthera.2016.03.013 |
| [3] |
K.M. Tevis, Y.L. Colson, M.W. Grinstaff, Adv. Biosys. 1 (2017) 1700083. DOI:10.1002/adbi.v1.10 |
| [4] |
T. Hagemann, S.C. Robinson, M. Schulz, L. Trumper, et al., Carcinogenesis 25 (2004) 1543-1549. DOI:10.1093/carcin/bgh146 |
| [5] |
T. Hagemann, J. Wilson, H. Kulbe, et al., J. Immunol. 175 (2005) 1197-1205. DOI:10.4049/jimmunol.175.2.1197 |
| [6] |
S. Joshi, A.R. Singh, M. Zulcic, et al., Mol. Cancer Res. 12 (2014) 1520-1531. DOI:10.1158/1541-7786.MCR-13-0682 |
| [7] |
K. Vinnakota, Y. Zhang, B.C. Selvanesan, et al., J. Cell. Physiol. 232 (2017) 3468-3480. DOI:10.1002/jcp.v232.12 |
| [8] |
X. Zhao, J. Qu, Y. Sun, et al., Oncotarget 8 (2017) 30576-30586. |
| [9] |
P. Lu, V.M. Weaver, Z. Werb, J. Cell Biol. 196 (2012) 395-406. DOI:10.1083/jcb.201102147 |
| [10] |
F. Spill, D.S. Reynolds, R.D. Kamm, et al., Curr. Opin. Biotechnol. 40 (2016) 41-48. |
| [11] |
F. Kai, H. Laklai, V.M. Weaver, Trends Cell Biol. 26 (2016) 486-497. DOI:10.1016/j.tcb.2016.03.007 |
| [12] |
M. Plodinec, M. Loparic, C.A. Monnier, et al., Nat. Nanotechnol. 7 (2012) 757-765. DOI:10.1038/nnano.2012.167 |
| [13] |
S. Su, Q. Liu, J. Chen, et al., Cancer cell 25 (2014) 605-620. DOI:10.1016/j.ccr.2014.03.021 |
| [14] |
K. Jin, N.B. Pandey, A.S. Popel, Oncotarget 8 (2017) 60210-60222. |
| [15] |
E. Cukierman, R. Pankov, D.R. Stevens, et al., Science 294 (2001) 1708-1712. DOI:10.1126/science.1064829 |
| [16] |
S. Ma, J. Zhou, Y. Zhang, et al., ACS Appl. Mater. Interfaces 8 (2016) 28468-28479. DOI:10.1021/acsami.6b09633 |
| [17] |
L. Gutzweiler, S. Kartmann, K. Troendle, et al., Biofabrication 9 (2017) 025027-025037. DOI:10.1088/1758-5090/aa7218 |
| [18] |
S. Breslin, L. O'Driscoll, Drug Discov. Today 18 (2013) 240-249. DOI:10.1016/j.drudis.2012.10.003 |
| [19] |
G. Lazzari, P. Couvreur, S. Mura, Polym. Chem. 8 (2017) 4947-4969. DOI:10.1039/C7PY00559H |
| [20] |
Z. He, W. Zhang, S. Mao, et al., Anal. Chem. 90 (2018) 5540-5545. DOI:10.1021/acs.analchem.8b00755 |
| [21] |
M. Jie, S. Mao, H. Li, et al., Chin. Chem. Lett. 28 (2017) 1625-1630. DOI:10.1016/j.cclet.2017.05.024 |
| [22] |
L. Lin, X. Lin, L. Lin, et al., Anal. Chem. 89 (2017) 10037-10044. DOI:10.1021/acs.analchem.7b02593 |
| [23] |
R. Li, J.D. Hebert, T.A. Lee, et al., Cancer Res. 77 (2017) 279-290. DOI:10.1158/0008-5472.CAN-16-0442 |
| [24] |
J. Bai, G. Adriani, T.M. Dang, et al., Oncotarget 6 (2015) 25295-25307. |
| [25] |
J.H. Lee, S.K. Kim, I.A. Khawar, et al., J. Exp. Clin. Cancer Res. 37 (2018) 4-15. DOI:10.1186/s13046-017-0654-6 |
| [26] |
M. Serra, I. Pereiro, A. Yamada, et al., Lab Chip 17 (2017) 629-634. DOI:10.1039/C6LC01319H |
| [27] |
A. Zuchowska, E. Jastrzebska, M. Chudy, et al., Anal. Chim. Acta 990 (2017) 110-120. DOI:10.1016/j.aca.2017.07.009 |
| [28] |
D.M. Richards, J. Hettinger, M. Feuerer, Cancer Microenviron. 6 (2013) 179-191. DOI:10.1007/s12307-012-0123-x |
| [29] |
F. Geissmann, M.G. Manz, S. Jung, M.H. Sieweke, et al., Science 327 (2010) 656-661. DOI:10.1126/science.1178331 |
| [30] |
Y. Komohara, M. Jinushi, M. Takeya, Cancer Sci. 105 (2014) 1-8. DOI:10.1111/cas.12314 |
| [31] |
P. Jeannin, L. Paolini, C. Adam, et al., FEBS J. 285 (2018) 680-699. DOI:10.1111/febs.2018.285.issue-4 |
| [32] |
E. van Overmeire, B. Stijlemans, F. Heymann, et al., Cancer Res. 76 (2016) 35-42. |
| [33] |
M. Miron-Mendoza, J. Seemann, F. Grinnell, Biomaterials 31 (2010) 6425-6435. DOI:10.1016/j.biomaterials.2010.04.064 |
| [34] |
J. Steinwachs, C. Metzner, K. Skodzek, et al., Nat. Methods 13 (2016) 171-176. DOI:10.1038/nmeth.3685 |
| [35] |
H. Ahmadzadeh, M.R. Webster, R. Behera, et al., Proc. Natl. Acad. Sci. U. S. A. 114 (2017) E1617-E1626. DOI:10.1073/pnas.1617037114 |
| [36] |
T. Lecuit, A.S. Yap, Nat. Cell Biol. 17 (2015) 533-539. DOI:10.1038/ncb3136 |
| [37] |
M. Canel, A. Serrels, M.C. Frame, et al., J. Cell Sci. 126 (2013) 393-401. DOI:10.1242/jcs.100115 |
| [38] |
J.F. Zhao, C. Klausen, X. Qiu, et al., Oncotarget 7 (2016) 28881-28890. |
| [39] |
J. Massague, Cell 134 (2008) 215-230. DOI:10.1016/j.cell.2008.07.001 |
| [40] |
H.Y. Chong, Y. Vodovotz, G.W. Cox, et al., J. Cell. Physiol. 178 (1999) 275-283. |
| [41] |
X.Z. Ye, S.L. Xu, Y.H. Xin, et al., J. Immunol. 189 (2012) 444-453. DOI:10.4049/jimmunol.1103248 |