 2019, Vol. 30
2019, Vol. 30 


b School of Physical Science and Technology (SPST), ShanghaiTech University, Shanghai 201210, China;
c University of Chinese Academy of Sciences, Beijing 100049, China
Large-scale combustion of non-renewable fossil fuels leads to the rapid growth of the concentration of atmospheric CO2, which gives rise to a series of problems [1-4]. Electrochemical reduction of CO2 using renewable energy to generate high value-added chemicals or fuel becomes an attractive strategy for utilization of CO2 resources [5]. A lot of efforts have been made to screen electrocatalysts that can reduce carbon dioxide efficiently, and inhibit the competitive hydrogen evolution reaction in the aqueous solution, such as oxidation treatment of metal electrodes including Au [6], Ag [7, 8], Sn [9], Pb [10], Co [11] and nanostructured optimization including nanoparticle [12], nanowire [13], nanoneedle [14] and nanoporous structure [15]. However, these efforts are limited to the conversion of carbon dioxide to generate carbon monoxide or formic acid through a two-electron pathway. The synthesis of electrocatalysts for production of higher value-added C2 & C2+ products, especially ethylene as an important monomer in polymer synthesis, is still a great scientific challenge [16].
Copper-based electrodes have been attracting extensive research efforts as copper has great potential to catalyze electrochemical CO2 reduction reaction (CRR) towards ethylene. CRRs on polycrystalline copper electrode mainly lead to mixtures of methane, ethylene, and carbon monoxide [17]. The partial current density for ethylene is only ca. 1.5 mA/cm2 with a low selectivity. Although copper single crystal electrode [18], copper electrode with rough surface [19] and copper nanocubic crystal [20] show improved faradaic efficiency for ethylene, the partial current density for ethylene is relatively low.
Recently, copper electrode covered with oxides exhibited excellent performance for ethylene formation. It has been proposed that copper oxide precursor was reduced to metallic copper with a unique microstructure under a negative potential environment during CO2 reduction reaction [21]. However, in-situ characterizations pointed to the existence of oxidized copper species even after the electrolysis process [22, 23]. A large number of Cu1+ sites might facilitate the stable adsorption of intermediate products and thus promote the selective generation of ethylene and other multi-carbon compounds [24].
Electrochemical treatment of copper in a KCl solution can also significantly improve the faradaic efficiency of C2H4 [25, 26]. The chloride ion presumably induced the formation of cubic particles on the copper surface with a large amount of exposed (100) facets, often considered as the active facet for ethylene production [27]. In addition, the chloride ion might stabilize the oxidized state and delay the reduction of cuprous oxide to metallic copper, which could promote the formation of reduction products with longer carbon chain [24].
Herein, we report the direct use of cuprous chloride as the catalyst precursor for electrochemical reduction of carbon dioxide. During the CRR, the precursor gradually turned into biphasic Cu2OCu cubes (CB-Cu), showing a relatively high faradaic efficiency and partial current density for the formation of ethylene, ethanol and other C2 & C2+ products.
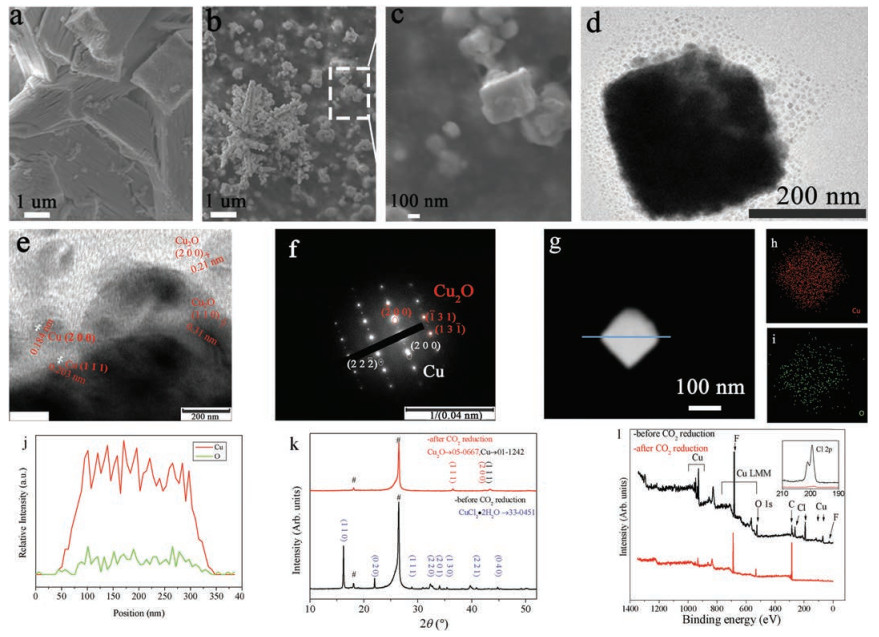
The electrode was prepared by printing a catalyst precursor ink uniformly onto a PTFE treated carbon paper (Supporting information). Large particles with the size of 1.5–2.5 μm were formed initially (Fig. 1a). XRD indicates that CuCl2·2H2O (eriochalcite) was formed with other unknown species during the electrode-preparation processes (Fig. 1k). After CRR for ca. 1 h, the large particles were transformed into smaller ones with the size of 150– 350 nm (Fig. 1b). Flower-like aggregates of these particles were observed. High-magnification SEM and TEM images (Figs. 1c and d) show the formation of cuboid particles. XRD indicates the presence of both copper and cuprous oxide (Fig. 1k). The high-resolution TEM image in Fig. 1e shows lattice fringe spacing of 0.21 nm and 0.31 nm, indexed to Cu2O (200) and (110) facet direction, respectively, as well as lattice fringe spacing of 0.184 nm and 0.203 nm corresponding to Cu (200) and Cu (111), respectively. SAED further confirms the existence of both copper and cuprous oxide (Fig. 1f). Scanning transmission electron microscopy equipped with energy dispersive X-ray spectroscopy (STEMEDS) shows that copper element distributes densely on the whole particle, while oxygen element is less dense (Figs. 1g–j). The combined results of STEM-EDS and HRTEM suggest a random mixture of copper and cuprous oxide at the shell of the cuboid particles.

|
Download:
|
| Fig. 1. Characterization of CuCl electrode. SEM images of (a) CuCl electrode before the electrolysis; (b, c) CuCl electrode at -1.06 V after 3680 s' electrolysis; TEM images of selected cuboid particle for CuCl after 3680 s' electrolysis; (d) low magnification; (e) high magnification; (f) SAED pattern; STEM-EDS images of selected cuboid particle for CuCl after 3680 s' electrolysis showing elemental distribution of (h) copper; (i) oxygen, and line-scan mode (j) (along line in (g)); X-ray diffraction (k) and XPS data (l) for CuCl electrode before and after 3680 s' electrolysis. The hashes in (k) corresponds to carbon substrate. F 1s peak in (l) results from carbon substrate with PTFE treated. | |
{kind=link}
No Cl element was detected by XPS after the CRR (Fig. 1l). Peaks at 935.4 and 955.4 eV detected for the electrode before CRR were attributed to Cu 2p3/2 and Cu 2p1/2 (Fig. S1 in Supporting information). Satellite peaks at 942.9 eV indicated the presence of Cu2+ [28].The broad peak of Cu 2p3/2 was fitted into two peaks with binding energy of 935.8 eV (major) and 933.2 eV (minor), corresponding to Cu2+ and Cu1+/0, respectively. Because the binding energy of Cu1+ and Cu0 is very close, it is very difficult to distinguish the two states based on Cu 2p3/2 data [29]. After the CRR, the characteristic peak of Cu2+ disappeared, and the Cu 2p peak moved to lower binding energy.These observations are also consistent with a depletion of Cl element and a formation of a biphasic Cu2OCu for the electrode during the CRR.
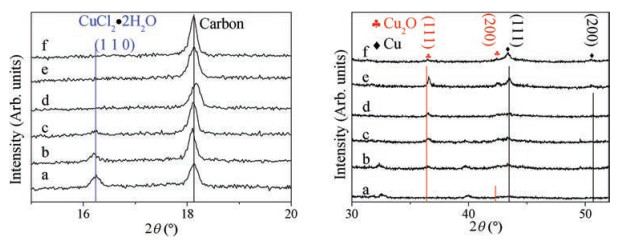
To further explore the changes of the morphology and the phase of the catalyst in the reaction process, the CuCl electrodes at six different reaction stages were compared by time resolved ex-situ XRD (Fig. 2) and SEM (Fig. 3). Intensities of the peaks corresponding to CuCl2·2H2O decreased gradually and virtually disappeared after 600 s' electrolysis. While those corresponding to copper and cuprous oxide increased as the CRR proceeded. When the reaction time was extended to 6 h, the intensity of (111) peak corresponding to Cu2O decreased relative to the Cu (111) peak, suggesting that the cuprous oxide was partially reduced to copper during a long period of CRR [24, 30]. The significantly different intensities of the peaks corresponding to Cu2O for the electrodes with different electrolysis time but same air exposure time strongly suggest that aerobic oxidation only cannot explain the observation of Cu2O and there should be Cu2O on the electrode even after a certain period of CRR. It can be seen from Fig. 3, the cubic crystals began to form after 60 s' CRR, and the electrode surface was covered with a large number of cubes with a size of 150–350 nm after electrolysis for 600 s. After a long period of electrolysis, the shape of the cubic crystal was still reserved, showing good morphological stability.

|
Download:
|
| Fig. 2. Time resolved ex-situ XRD patterns of CuCl electrode during the electrolysis at -1.06 V. (a) CO2 was purged for 30 min, (b) after LSV, (c) 60 s, (d) 600 s, (e) 3680 s, and (f) 6 h after the start of the CRR. | |
{kind=link}

|
Download:
|
| Fig. 3. Time resolved ex-situ SEM images of CuCl electrode at -1.06 V. (a) CO2 was purged for 30 min, (b) after LSV, (c) 60 s, (d) 600 s, (e) 3680 s, and (f) 6 h after the start of CRR. | |
{kind=link}
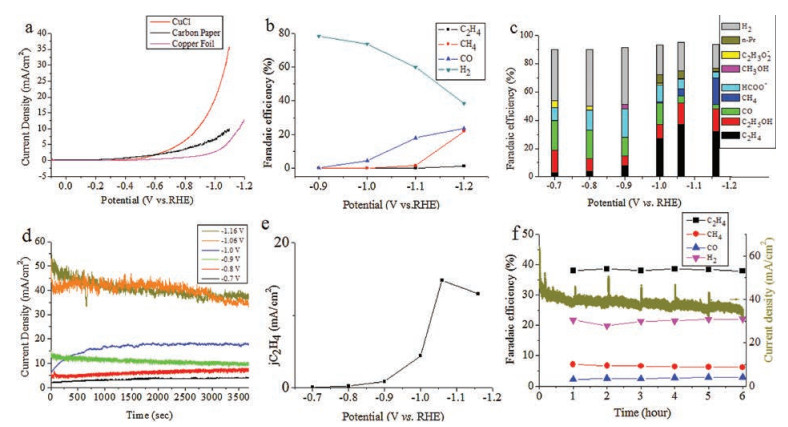
The potentiostatic electrolysis measurement was carried out in a CO2 saturated 0.1 mol/L KHCO3 solution. Gas and liquid products were detected by gas chromatography and nuclear magnetic resonance, respectively. LSV was used to compare the overall activity of the CuCl electrode, the carbon paper substrate and the copper foil. As shown in Fig. 4a, the catalyst led to significant increase of the current density. For carbon paper substrate, hydrogen evolution dominated at -0.9 V. The onset potential of methane was -1.1 V, while ethylene emerged at -1.2 V with faradaic efficiency of only 1.2%. We investigated the electrochemical activity of CB-Cu to reduce carbon dioxide under a series of potentials. As shown in Fig. 4c, under the low overpotential (-0.7 V~-0.9 V), the gaseous products were dominated by H2 and CO, with faradaic efficiency of 40% and 21%, respectively. Ethanol was the main C2 & C2+ product. A considerable amount formate formation (FE: 9%–20%) was also formed in the liquid phase, as well as a small amount of methanol and acetate (FE ≤ 5%). With increasing overpotential (-0.9 V~-1.16 V), the FE of H2, CO, and formate decreased. At -1.16 V, the minimum FE of H2 was reached (17%), and the overall selectivity for CRR was 76.5%.

|
Download:
|
| Fig. 4. Catalytic behavior for electroreduction of CO2 on CB-Cu. (a) LSV comparison for CB-Cu electrocatalyst, carbon paper and the copper foil, (b) gaseous products distribution on carbon paper, (c) faradaic efficiency of products on CB-Cu, (d) current densities as a function of time at different applied potentials, (e) partial current density of ethylene as a function of applied potentials, (f) 6-h stability performance of CB-Cu at -1.06 V vs. RHE. | |
{kind=link}
When the potential shifted from -0.9 V to -1 V, the FE of ethylene increased 3.4 times, and n-propanol and methane started to appear. At -1.06 V, a maximum ethylene FE of 37% was obtained and the overall C2 & C2+ products selectivity reached 57.5%, including ethanol FE of 15%, n-propanol FE of 5%, and acetate FE of 0.5%. The selectivity of the C2 & C2+ products (Fig. S6 in Supporting information) is at the same level with that obtained using the Cu2O electrode as the starting catalytic material [25, 31, 32]. When the potential was negatively shifted to -1.16 V, the faradaic efficiency of ethylene decreased slightly, which may be related to limited mass transfer of carbon dioxide [33].
Cl in the electrolyte is known to affect activity and selectivity of CO2 electroreduction on Cu catalysts [34, 35]. Control experiment was carried out by replacing the electrolyte for the activation of the chloride catalyst precursor with freshly prepared electrolyte during a continuous electrolysis (SI), and only little change was observed on current density and product selectivity, suggesting that the chloride dissolved in the original electrolyte is probably not important in the electrolysis. In comparison, the presence of Cl was proposed to delay the reduction of Cu2O [24], which might facilitate the formation of C2+ products. The high selectivity of ethylene may benefit from the in-situ formation of biphasic Cu2OCu induced by Cl [36] during the reaction. Recent studies have shown that the interface between Cu1+ and Cu0 contributes to the dimerization of CO adsorbed on the electrode surface to generate ethylene [37]. In addition, Yang et al. [38] emphasized the role of the structural transformation of Cu nanoparticles into cube particles during the reaction to increase the selectivity of the C2 & C2+ product. The cubic mesocrystals were formed during the present in-situ reduction process, which may also contribute to the improved ethylene selectivity [25, 26].
Fig. 4d showed that the current density increased from 3.35 mA/cm2 to 40.51 mA/cm2 with a negative shift in the applied potential. As shown in Fig. 4e, CB-Cu produced a maximum ethylene current density of 14.8 mA/cm2 at -1.06 V and showed fairly good stability in the 6-h test (Fig. 4f).
Table S1 (Supporting information) compares ethylene production performance of CB-Cu catalyst with other copper-based catalysts in the literature. As for the partial current density of ethylene, CB-Cu was comparable or better than the reported catalysts. We tentatively attribute this enhancement to the increased surface roughness of our chloride-derived copper compared to the oxide-derived copper reported previously.
In summary, we studied the catalytic activity of a CuCl-based electrode for electrochemical reduction of carbon dioxide and found that CuCl was gradually transformed into a cubic structure of biphasic Cu2O-Cu presumably due to the depletion and induction of chlorine ions in the reduction process. Enhanced faradaic efficiency and partial current density for the formation of ethylene were observed for this new catalyst with a good stability. The synergy of Cu1+ and cubic structure may account for this improvement. The utilization of cuprous chloride as a catalyst precursor in replace of cuprous oxide provides a new strategy to improve the selectivity of C2 & C2+ products.
AcknowledgmentsThis work was financially supported by Shell-CAS Frontier Sciences Program (No. PT48809) from Shell and start-up funding from ShanghaiTech University.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.07.003.
| [1] |
M. Liu, T. Qin, Q. Zhang, et al., Sci. China:Chem. 58 (2015) 1524-1531. DOI:10.1007/s11426-015-5405-y |
| [2] |
S. Chu, A. Majumdar, Nature 488 (2012) 294-303. DOI:10.1038/nature11475 |
| [3] |
M.S. Dresselhaus, I.L. Thomas, Nature 414 (2001) 332-337. DOI:10.1038/35104599 |
| [4] |
A. Chen, B.L. Lin, Joule 2 (2018) 594-606. DOI:10.1016/j.joule.2018.02.003 |
| [5] |
X.F. Bai, W. Chen, B.Y. Wang, et al., Acta Phys. Chim. Sin. 33 (2017) 2388-2403. |
| [6] |
Y. Chen, C.W. Li, M.W. Kanan, J. Am. Chem. Soc. 134 (2012) 19969-19972. DOI:10.1021/ja309317u |
| [7] |
M. Ma, B.J. Trzesniewski, J. Xie, et al., Angew. Chem. Int. Ed. 55 (2016) 9748-9752. DOI:10.1002/anie.201604654 |
| [8] |
H. Mistry, Y.W. Choi, A. Bagger, et al., Angew. Chem. Int. Ed. 56 (2017) 11394-11398. DOI:10.1002/anie.201704613 |
| [9] |
S. Zhang, P. Kang, T.J. Meyer, J. Am. Chem. Soc. 136 (2014) 1734-1737. DOI:10.1021/ja4113885 |
| [10] |
C.H. Lee, M.W. Kanan, ACS Catal. 5 (2015) 465-469. DOI:10.1021/cs5017672 |
| [11] |
S. Gao, Y. Lin, X. Jiao, et al., Nature 529 (2016) 68-71. DOI:10.1038/nature16455 |
| [12] |
D. Gao, H. Zhou, J. Wang, et al., J. Am. Chem. Soc. 137 (2015) 4288-4291. DOI:10.1021/jacs.5b00046 |
| [13] |
W. Zhu, Y. J. Zhang, H. Zhang, et al., J. Am. Chem. Soc. 136 (2014) 16132-16135. DOI:10.1021/ja5095099 |
| [14] |
M. Liu, Y. Pang, B. Zhang, et al., Nature 537 (2016) 382-386. DOI:10.1038/nature19060 |
| [15] |
Q. Lu, J. Rosen, Y. Zhou, et al., Nat. Commun. 5 (2014) 3242-3247. DOI:10.1038/ncomms4242 |
| [16] |
D.D. Zhu, J.L. Liu, S.Z. Qiao, Adv. Mater. 28 (2016) 3423-3452. DOI:10.1002/adma.201504766 |
| [17] |
K.P. Kuhl, E.R. Cave, D.N. Abram, et al., Energy Environ. Sci. 5 (2012) 7050-7059. DOI:10.1039/c2ee21234j |
| [18] |
Y. Hori, I. Takahashi, O. Koga, et al., J. Phys. Chem. B 106 (2002) 15-17. DOI:10.1021/jp013478d |
| [19] |
W. Tang, A.A. Peterson, A.S. Varela, et al., Phys. Chem. Chem. Phys. 14 (2012) 76-81. DOI:10.1039/C1CP22700A |
| [20] |
A. Loiudice, P. Lobaccaro, E.A. Kamali, et al., Angew. Chem. Int. Ed. 128 (2016) 5789-5792. |
| [21] |
S. Lee, J. Lee, Chemsuschem. 9 (2016) 333-344. DOI:10.1002/cssc.v9.4 |
| [22] |
D. Kim, S. Lee, J.D. Ocon, et al., Phys. Chem. Chem. Phys. 17 (2015) 824-830. DOI:10.1039/C4CP03172E |
| [23] |
H. Mistry, A.S. Varela, C.S. Bonifacio, et al., Nat Commun. 7 (2016) 12123-12130. DOI:10.1038/ncomms12123 |
| [24] |
S. Lee, D. Kim, J. Lee, Angew. Chem. Int. Ed. 54 (2015) 14701-14705. DOI:10.1002/anie.201505730 |
| [25] |
C.S. Chen, A.D. Handoko, J.H. Wan, et al., Catal. Sci. Technol. 5 (2015) 161-168. DOI:10.1039/C4CY00906A |
| [26] |
F.S. Roberts, K.P. Kuhl, A. Nilsson, Angew. Chem. Int. Ed. 127 (2015) 5268-5271. DOI:10.1002/ange.v127.17 |
| [27] |
K.J. Schouten, Z. Qin, E. P. Gallent, et al., J. Am. Chem. Soc. 134 (2012) 9864-9867. DOI:10.1021/ja302668n |
| [28] |
A. Dutta, M. Rahaman, M. Mohos, et al., ACS Catal. 7 (2017) 5431-5437. DOI:10.1021/acscatal.7b01548 |
| [29] |
N.S. Mcintyre, M.G. Cook, Anal. Chem. 47 (1975) 2208-2213. DOI:10.1021/ac60363a034 |
| [30] |
P.D. Luna, R. Quinterobermudez, C.T. Dinh, et al., Nat. Catal. 1 (2018) 103-110. DOI:10.1038/s41929-017-0018-9 |
| [31] |
A. Dutta, M. Rahaman, N.C. Luedi, et al., ACS Catal. 6 (2016) 3804-3814. DOI:10.1021/acscatal.6b00770 |
| [32] |
D. Gao, I. Zegkinoglou, N.J. Divins, et al., ACS Nano. 11 (2017) 4825-4831. DOI:10.1021/acsnano.7b01257 |
| [33] |
C.W. Li, J. Ciston, M.W. Kanan, Nature 508 (2014) 504-507. DOI:10.1038/nature13249 |
| [34] |
A.S. Varela, W. Ju, T. Reier, et al., ACS Catal. 6 (2016) 2136-2144. DOI:10.1021/acscatal.5b02550 |
| [35] |
D. Gao, F. Scholten, B.R. Cuenya, ACS Catal. 7 (2017) 5112-5120. DOI:10.1021/acscatal.7b01416 |
| [36] |
H. Liu, Y. Zhou, S.A. Kulinich, et al., J. Mater. Chem. A. 1 (2013) 302-307. DOI:10.1039/C2TA00138A |
| [37] |
H. Xiao, W.A. Goddard, T. Cheng, Y. Liu, Proc. Natl. Acad. Sci. U. S. A. 114 (2017) 6685-6688. |
| [38] |
D. Kim, C.S. Kley, Y. Li, et al., Proc. Natl. Acad. Sci. U. S. A. 114 (2017) 10560-10565. DOI:10.1073/pnas.1711493114 |