 2019, Vol. 30
2019, Vol. 30 


b The Scientific Research Center, China-Japan Union Hospital, Jilin University, Changchun 130033, China
During the past 30 years, quantum dots (QDs), containing hundreds to thousands of atoms, have drawn significant scientific interest due to their unique size-dependent chemical and physical properties, different from those of molecular or bulk materials [1-7]. Among the whole QD-family, metal selenide quantum dots (MSQDs) and metal telluride quantum dots (MTQDs) have been extensively studied and shown great potential for practical applications in broadening areas. To name just the representative ones, CdSe and CdTe QDs with full emission from blue to red, have been used as optimal emitting materials for lighting and display [8-11]. Owning to the intense surface plasmon resonance effect under near-infrared light irradiation, copper-containing selenide and/or telluride QDs have shown excellent performances in photothermal therapy by converting light to heat [12-17]. PbTe and SnTe QDs have been identified as ideal thermoelectric materials with high heat-to-electricity conversion efficiencies [18-22]. With good biocompatibility and permeability, ZnSe and Mn:ZnSe QDs have been successfully tested for bioimaging [23-28].
Owning to the board applications in diversified fields, great efforts have been devoted to exploiting the chemically synthetic methods for MSQDs and MTQDs to meet the growing practical demands, including the solvothermal method, sonochemical method, electrochemical method, biochemical method, and colloidal chemical method. The solvothermal method is featured for the high pressure and temperature with less control over the size distribution and surface state of the resulting QDs [29-33]. The sonochemical method causes the nucleation and growth of QDs through acoustic cavitation by generating hotspot in implosive bubbles [34-37]. The sonochemical method can yield smaller and more uniformed QDs than the solvothermal method without exogenous heating, but requires high-intensity ultrasonic probe equipment. The electrochemical method provides an electric current pulse to initialize the nucleation and electrodeposition, followed by an exfoliation process from the probe electrodes [38-42]. Although the electrochemical route is convenient and can be operated at room temperature, aggregation or stacking always occurs in the resulting QDs. The biochemical method is performed in living organisms. Through metal detoxification pathway, watersoluble QDs can be produced after feeding the biosome with raw materials [43-46]. The biochemical method seems straightforward and environmentally benign, but the type, yield and quality of QDs are rather limited. The colloidal chemical method involves the reactions between the precursors in the solvent with capping ligands, which can be tuned by various parameters [47-50]. Even though the adjustment rule varies and seems complicated in different systems, the colloidal chemical method is the mainstream to produce functional MSQDs and MTQDs, affording powerful control over the composition, monodispersity, shape and surface state.
Thus far, colloidal synthesis of MSQDs and MTQDs has been reviewed in many previous reports [51-53]. Additionally, numerous reviews have focused on nucleation-growth mechanism, structure characterization, compositional modulation, shape engineering, property tailoring, and specific applications [54-59]. In contrast to the previous works, we focus on the development of green synthetic methodologies, in view of the raw materials, energy- and time-consumption, byproducts and waste, and simplicity of the artificial process. For the sake of clarity, in this comprehensive review, the colloidal chemical method is categorized into two systems according to the polarity of the reaction solvent: the organic phase system and the aqueous phase system. With the development of the precursors as the mainline, we summarize the recent advances in green methods for MSQDs and MTQDs, highlighting the phosphine-free precursors and aqueous ionic precursors.
2. Organic phase systemThe colloidal chemical method in organic phase system for MSQDs and MTQDs can be traced back to 1988. Brus and coworkers used organosilylselenide to passivate Cd-rich micelle stabilized CdSe crystallites and obtained stable and soluble CdSe QDs [60]. Later in 1993, an innovative synthetic route to prepare MSQDs and MTQDs was put forward by Bawendi's group [61]. They selected dimethyl cadmium (Me2Cd) dissolved in trioctylphosphine oxide (TOPO) as the metal source and selenium or tellurium dissolved in trioctylphosphine (TOP) as the chalcogen source. Both the organometallic and chalcogen sources were injected into hot TOPO solution at the same time and nearly monodisperse CdSe and CdTe QDs were gained. The synthetic apparatus is shown in Scheme 1. This hot-injection mode is still the most classic and developed in synthesizing MSQDs and MTQDs so far. Nevertheless, Me2Cd, as the Cd source, is extremely toxic, unstable and explosive, the use of which requires strictly anhydrous and anaerobic conditions and equipment. Large-scale synthesis is hardly possible for this reason either. As a result, this actuality limited the Me2Cdrelated schemes in only a few research groups at that time. In 2001, Peng's group demonstrated that Me2Cd was not essential in the synthesis process [62]. Me2Cd was identified to decompose in hot TOPO and convert to cadmium phosphate complex in the presence of hexylphosphonic acid (HPA) or tetradecylphosphonic acid (TDPA). Experimentally, they replaced Me2Cd by cheap and stable CdO as the Cd precursor. With TOPO as the coordinating solvent, HPA or TDPA as the ligand and selenium or tellurium powders dissolved in tributylphosphine (TBP) as the chalcogen precursor, Peng's group synthesized high-quality CdSe and CdTe QDs under milder conditions reproducibly. This new scheme permitted largescale synthesis of MSQDs and MTQDs in a glove-box and achieved initial understanding of their nucleation and growth steps, which was a landmark in the development of synthetic methods. Thereafter, various cheap and simple inorganic metal slats or metal oxides began to be used as metal precursors [63-66]. Diverse ligands, such as oleic acid (OA), oleylamine (OLA), 1-dodecanethiol (DT), octadecylphosphonic acid (ODPA) and so forth, were also exploited [67-71]. In addition, non-coordinating solvents, including octadecene (ODE), paraffin and benzyl ether (BE) were also introduced to replace coordinating TOPO [72-74]. By adjusting the above mentioned parameters, high-quality MSQDs and MTQDs could be obtained with desired shapes and properties. Despite the great progress in the synthetic schemes, the monotonous approach to access chalcogen precursors is the dissolution of selenium or tellurium powders in organophosphorus, such as TOP and TBP, which are expensive, air-sensitive and environmentally pollutional [61, 75, 76]. Obviously, it is significant to replace the organophosphorus by any chemicals with less cost, sensitivity or toxicity, which can be termed as "green method", or at least, as "greener method".

|
Download:
|
| Scheme 1. Schematic illustration of the synthetic apparatus for hot-injection mode. | |
{kind=link}
2.1. Single-source precursor
To avoid the use of organophosphorus, an alternative approach with a single-source precursor was attempted by several groups. In 1996, Trindade and O'Brien obtained crystalline CdSe QDs from a single-source precursor decomposition for the first time by injecting methyl diethyldiselenocarbamato cadmium(Ⅱ) (MeCddsc: [CH3CdSe2CN(C2H5)2]2) into TOPO at 250 ℃ [77]. Later, O'Brien's group studied the procedure detailedly and used 2, 2- bipyrimidine to assemble the CdSe QDs into composites [78]. By tailoring appropriate single-source precursors, this strategy was applied for the synthesis of diverse MSQDs [79-81]. Despite the extension of this method, the alkylcadmium or alkylzinc diseleno carbamate complex is air-sensitive, which needs air-free manipulations and increases the complexity of the synthetic process. In this scenario, simple air-stable methylhexyldiselenocarbamato of cadmium and zinc complex precursors were further designed [82]. The synthetic method using a single-source precursor seems relatively safe and straightforward. However, it requires extra processes to prepare the complex precursor containing both the metal and chalcogen, which always involves expensive and toxic reagents. In addition, the metal-to-chalcogen molar ratio is fixed at such a certain value that cannot be simply adjusted. Noteworthily, MTQDs are scarcely synthesized with a single-source precursor, since it is difficult to prepare a complex precursor composed of metal and tellurium. Therefore, phosphine-free synthetic method with the single-source precursor is less developed than that with the separate precursors.
2.2. Phosphine-free Se precursorDue to the relatively low melting point at 221 ℃, selenium powders can be directly dissolved in various organic high-boilingpoint solvents under heating. In 2005, Mulvaney's group heated a mixture of selenium powder and ODE up to 200 ℃ under nitrogen atmosphere and obtained a transparent Se-solution [83]. Taking the advantage of the hot-injection mode, they synthesized nearly monodispersed CdSe QDs with the resulting phosphine-free precursor and also demonstrated that the structure of CdSe QDs could be alerted by the variation of ligand types. In the same year, Zou and coworkers dissolved selenium powder in hot paraffin as the phosphine-free Se precursor and swiftly injected the Cd-OA solution into the homogeneous Se solution at 220 ℃ [84]. Through this approach, high-quality CdSe QDs were also gained. Note that the reduction of selenium to H2Se gas by long alkane chain solvents was identified for the first time. Based on such dissolution mechanism, selenium powder was therewith dissolved in similarly structured high-boiling solvents, such as OLA, N, N-dimethyloleoylamide, olive oil, N-oleoylmorpholine and so forth [85-88]. Peng's group also proved that Se-ODE suspension (Se-SUS) could act as powerful phosphine-free precursor for producing binary, coreshell and doped metal selenide QDs [89, 90]. Obviously, the Se-SUS is more facile without the generally tedious heating step, but the heterogeneous solution offers less control over the nucleation process of MSQDs.
In 2005, Cao's group simplified the hot-injection mode to produce MSQDs and achieved CdSe QDs with a non-injection mode, where they directly heated the mixture of selenium powder, cadmium myristate and ODE with subsequent addition of OA under nitrogen atmosphere [91]. Soon after, they employed cheaper selenium dioxide to replace selenium as the precursor and synthesized CdSe QDs in the open air [92]. Based on the noninjection mode, ternary CdSeS QDs and magic sized CdSe QDs were also synthesized by Yu and coworkers [93-96]. This non-injection mode is straightforward and suitable for large-scale industrial production of MSQDs. However, imbalance between the nucleation and growth steps is the main drawbacks of the protocol.
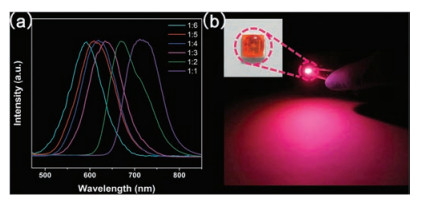
Except for the direct long heating approach, phosphine-free Se precursors can be prepared by dissolving selenium powder in organic solvents at room-temperature with the assistance of reductants. In 2010, Ying's group employed sodium borohydride (NaBH4) to reduce element selenium to Se2- and facilitate its dissolution in OLA under sonication [97]. This phosphine-free Se precursor is highly reactive and applicable to synthesize various MSQDs, including CdSe, PbSe, ZnSe, CuInSe2 and CdSe/CdSe QDs. NaBH4 plays a critical role in the process, without which, selenium powder can be dissolved in OLA only after extended heating. However, due to the excessively used NaBH4, impurities, such as boron and alkali metal ions, are introduced into the reaction system, which may influence the reaction mechanism and subsequent separation process. In addition, NaBH4 is not an ideal reducing agent for its easy oxidation in air. In 2012, Zhang's group developed a facile method for preparing Se precursor by dissolving selenium powder in a mixture of DT and OLA ambiently [98]. Selenium powder cannot be dissolved in either OLA or DT alone at room-temperature (Fig. 1a). In a mixture of the two, elemental selenium is reduced by DT into low valence Se and associates with OLA to form a soluble Se-OLA complex, whereas DT is oxidized into disulfides (Fig. 1b). The phosphine-free Se precursor is rather stable, and can even recover from oxidation state by the addition of extra DT after long-term open air storage (Fig. 1c). Additionally, the phosphine-free Se precursor is suitable for synthesis of a variety of MSQDs, including binary, ternary and even quaternary selenides. Due to the high reactivity, ultrasmall ternary Ag-In-Se alloy QDs can be achieved at relatively low temperature in a short duration, such as 88 ℃ and 150 s, which largely shortens the energy- and time-consumption in the synthesis process [99]. Moreover, the easily acquired Ag-In-Se alloy QDs cover the emission range from 592 nm to 716 nm with PLQY up to 9.9%, which exhibit great potentials in lighting (Fig. 2). The seminal phosphine-free precursor is safe, economical and suitable for rapid synthesis of MSQDs.

|
Download:
|
| Fig. 1. (a) Photograph of vials containing 1.5 mmol of Se powder in 1.5 mL of OLA, 1.5 mL of DT, and a mixture of 0.75 mL each of OLA and DT. (b) Equation for the reaction between Se powder and DT in the presence of OLA. (c) Entire redox cycle of the OLA/DT/Se system. The initial solution contained 0.0125 mmol of Se, 1.5 mL of OLA, and 3.2 μL of DT. Reprinted with permission [98]. Copyright 2012, American Chemical Society. | |
{kind=link}

|
Download:
|
| Fig. 2. (a) Spectra of the Ag–In–Se QDs that are synthesized by altering the Ag/In molar feed ratio from 1/1 to1/6. (b) LED prototype based on Ag–In–Se QDs with red emission. Inset: the photograph of LED taken under sunlight. Reproduced with permission [99]. Copyright 2015, Royal Society of Chemistry. | |
{kind=link}
2.3. Phosphine-free Te precursor
In contrast to selenium, elemental tellurium cannot be directly dissolved in similarly structured non-coordinating solvents under heating, due to its higher melting point and stronger metallicity. Thus, the phosphine-free method for Te precursor is less reported. In 2005, Fang and coworker obtained TeH- anion by introducing superhydride into the Te-TOP system under nitrogen atmosphere [100]. Later, Rajh's group prepared a Te precursor by mixing TeTOP, superhydride and OLA, and identified the presence polytellurides [101]. The active polytelluride precursor was successfully used to synthesize controllable ZnTe QDs by varying the precursor concentration, reaction time and reaction temperature. Although the polytelluride precursors are still based on the organophosphorus, they lead to a new thinking about stronger reduction method for further phosphine-free precursors.
In 2010, Li and coworkers developed an innovative method for synthesis of MTQDs. They prepared Te precursors by directly heating TeO2 in TOPO at 380 ℃ for 5 h and employed OA as the capping ligand [102]. TeO2 is identified to be reduced to Te0 and prefers to be dissolved in impure TOPO because of the presence of octylphosphinic acid (DOPA). The formation of a Te-DOPA complex promotes the Te0 dissolution in TOPO. This method bypasses the utilization of phosphines and is capable to control the structure and shape of the MTQDs, but the preparation of the Te precursor is too time- and energy-consuming to apply in practical manufactures. Besides, in 2012, Gupta's group adopted strong reducing agent, NaBH4, to enable the dissolution of tellurium powder in trioctylamine (TOA), which is similarly structured to TOP [103]. Binary Cr2Te3 and ternary CuCr2Te4 QDs were synthesized with the phosphine-free Te precursor and related magnetic properties were further explored. With the assistance of NaBH4, the reaction temperature to prepare the phosphine-free Te precursor is lowered down to 350–355 ℃, which is still severely energy-consuming.
Inspired by the phosphine-free method by Gupta's group, Zhang and coworkers employed NaBH4 to reduce tellurium powder in alkylamides for preparing Te precursor [104]. Te powders can be rapidly dissolved under mild condition, such as 80 ℃ for 5 min. Unique amide groups in the alkylamides, such as N, N-dimethylformamide (DMF), N, N-dimethylacetamide (DMAC) and N, N-diethylpropionamide (DEP), lead to larger polarity and permittivity than OLA or TOA, thus promoting the reduction of Te and stabilizing the reduced Te2- (Fig. 3a). Related sequence of the reaction is shown in Fig. 3b. This phosphine-free Te precursor is so reactive that can produce high-quality CdTe QDs at low temperatures in extremely short duration, such as 80 ℃ for 10 s. Once the Te precursor solution was injected into the flask containing the metal precursor solution, the heating source was immediately removed and the reaction flask was transferred into icy water to stop the reaction. By gradually elevating the temperature, sizes and optical properties of the CdTe QDs can be easily adjusted (Fig. 3c). It is undemanding to obtain ultralarge CdTe QDs just by prolonging the heating duration. Note that CdTe QDs can be prepared with the Te precursor even at room-temperature, despite the tedious reaction time. The phosphine-free Te precursor is validated to be suitable in synthesizing diverse MTQDs, but due to its ultrahigh reactivity, specialists are required to produce desired MTQDs with good controllability.

|
Download:
|
| Fig. 3. (a) The photographs in exhibit the dissolution of 1.5 mmol Te powder in 1.5 mL DMF, DMAC and DEP by heating the mixtures at 80 ℃ in the presence of 1.5 mmol NaBH4, and the mixture of 1.5 mmol Te powder and 1.5 mL DEP at 180 ℃ in the absence of NaBH4. (b) The proposed sequence of the reaction in producing soluble tellurium precursors by NaBH4 reduction in alkylamides. (c) The corresponding optical spectra and PL images of the CdTe QDs obtained at different temperatures. Reprinted with permission [104]. Copyright 2013, Royal Society of Chemistry. | |
{kind=link}
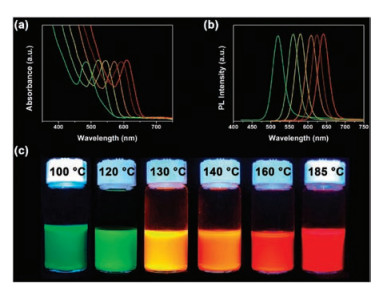
Recently, Zhang's group dissolved TeO2 in DT under mild heating to prepare moderately reactive phosphine-free Te precursor [105]. In the dissolution reaction, TeO2 is reduced to Te0 nanoclusters, whereas DT is oxidized into disulfides. Disulfides and excess DT interact with the Te0 nanoclusters through non-binding interactions to generate a stable colloidal Te solution. These nonbinding interactions are weak and can be disturbed by the addition of coordinating OLA, thus generating Te0 precipitates. By injecting this phosphine-free Te precursor into a Cd-OLA solution at different temperatures and maintaining the temperature for 10 min, brightly emissive CdTe QDs can be gained, covering the region from green to red (Fig. 4). With applicability for numerous MTQDs, this cheap, environmentally benign and stable Te precursor shows great potential for industrial production.

|
Download:
|
| Fig. 4. UV–vis absorption (a) and PL emission (b) spectra of the CdTe QDs that are synthesized by altering the reaction temperature respectively at 100, 120, 130, 140, 160 and 185 ℃. The corresponding PL images excited by 365 nm UV light are shown in (c). Reprinted with permission [105]. Copyright 2017, American Chemical Society. | |
{kind=link}
3. Aqueous phase system
Compared with organic phase system, the colloidal aqueous synthesis of MSQDs and MTQDs is advantageous for the environmentally benign and biocompatible solvent and mild synthetic conditions, which is closer to the "green chemistry" concept [106, 107]. Additionally, the resulting water-soluble QDs can be applied in bio-science directly [23, 59]. In the aqueous phase system, the metal precursors are generally common inorganic salts, whereas small organic molecules with binding groups, such as thiol, hydroxyl, carboxyl and amino, anchor to the surface of the QDs and play the role of protector and stabilizer [52]. The chalcogen precursor in aqueous phase system can be either anionic or cationic.
3.1. Anionic chalcogen precursorIn 1989, Henglein and coworkers obtained a Na2Te solution by bubbling the H2Te stream, generated in the reaction of Al2Te3 and H2SO4 through an icy NaOH solution [108]. On the basis of this protocol, NaHSe and NaHTe solutions were prepared as chalcogen precursors by passing adjusted amount of hydrogen chalcogenide through the NaOH solution. In 1993, Nozik's group mixed the freshly prepared NaHTe with Cd2+ solution in the presence of the 3-mercapto-l, 2-propanediol as the stabilizer and hexametaphosphate (HMP) as the buffer [109]. This mixed solution, termed as the CdTe precursor solution, was then refluxed in the open air to form CdTe QDs. Analogously, Eychmüller's group synthesized CdSe QDs by employing the NaHSe solution as the aqueous Se precursor [110]. To simplify the synthetic process, H2Se and H2Te gases were directly used as chalcogen precursors. Sheavel and coworkers passed H2Se gas through a mixture solution consisting of Zn2+ and thiol stabilizers to form the ZnSe precursor solution, and then refluxed the ZnSe precursor solution to promote the growth of ZnSe QDs [111]. CdTe QDs stabilized by different thiols, such as 1-thioglycerol, 2-mercaptoethanol, thioglycolic acid, 2-dimethylamino thanethiol, and 2-mercaptoethyl amine, were also synthesized with H2Te by Rogach and coworkers [112]. Note that pH of the CdTe precursor solution was adjusted to a certain value according to the thiol types before the reflux step. Weller's group directly passed H2Te gas through a N2 saturated aqueous solution containing HgClO4 and thiolglycerol at pH 11.2, and obtained HgTe QDs on a gram scale at room-temperature [113]. By employing mixed gases, such as H2S/H2Se, H2S/H2Te, H2Se/H2Te, ternary metal chalcogenide QDs were also prepared [114]. Scheme 2 presents the standard apparatus for the formation of the hydrogen chalcogenide gases and growth of MTQDs promoted by mild reflux [52]. These chalcogen precursors originated from hydrogen chalcogenides possess intrinsic properties of high toxicity and extreme air-sensitivity. Besides, it is hardly possible to strictly control the amount of hydrogen chalcogenide gases in the reaction. These drawbacks stimulate new researches for alternative anionic chalcogen precursors for corresponding QDs.

|
Download:
|
| Scheme 2. Presentation of the synthesis of aqueous thiol-capped MTQDs using hydrogen chalcogenides (H2Se, H2Te) as the chalcogen source. Reprinted with permission [52]. Copyright 2013, Royal Society of Chemistry. | |
{kind=link}
Early in 1969, Lalancette and Arnac adopted elemental chalcogens to replace hydrogen on borohydrides, generating reductive borohydrides NaBH2Se3 and NaBH2Te3 [115]. Taking the advantage of this scenario, Yang's group prepared a NaHSe solution for the first time by mixing borohydride and selenium in icy water with a molar ratio of 2:1, thus bypassing the requirement of toxic H2Se [116]. With the NaHSe solution as the Se precursor, mercaptopropionic acid (MPA) capped ZnSe and Cu-doped ZnSe QDs were further synthesized after a following reflux step. In 2003, Zhang et al. employed NaBH4 to reduce tellurium in icy water and gained a transparent NaHTe solution [117]. During the reaction, white byproduct precipitations of sodium tetraborate formed on the bottom of the flask and the NaHTe supernatant could be easily separated by a syringe. Colloidal CdTe solutions were prepared by adding the freshly prepared NaHTe solution into N2-saturated CdCl2 solutions at pH 9.0 in the presence of mercaptocarboxylic acids. The molar ratio of Cd2+/NaHTe/mercaptocarboxylic acids is 1/0.5/2.4. Then, the resulting mixture solution underwent a reflux step for different durations to obtain CdTe QDs with desired sizes. Though the reduction of both selenium and tellurium by NaBH4 in water at the same time, NaHSexTe1-x solutions could be also obtained [118]. The NaHSexTe1-x solutions were used as binary chalcogen precursors to synthesize complicated metal chalcogenide QDs. Because the NaBH4-based preparation routes to aqueous chalcogen precursors are non-hazardous and implementable, the resulting chalcogen precursors are widely used to synthesize MSQDs and MTQDs with various compositions, such as CdSe, CdTe, ZnSe, HgTe, CdHgTe, ZnSeS and so forth [52]. On the basis of the Te precursor, Zhang et al. prepared CdTe QD-polymer composites by adopting polymerizable surfactants as both the ligands and comonomers [119, 120]. Anisotropic 1D CdTe nanorods and nanowires were also fabricated by adjusting the types of thiol ligands [121, 122]. Besides, Zhang et al. found that the growth rate of the aqueous QDs could be accelerated by decreasing the electrostatic repulsion in the solution [123, 124]. On the basis of such understanding, Han et al. added NH3 or N2H4 into the synthetic system to promote the growth rate of the aqueous CdTe QDs, which was a combination of kinetic agglomeration growth and thermodynamic diffusion equilibrium [125]. The addition of NH3 or N2H4 facilitates the agglomeration growth by weakening the electrostatic repulsion between Cd monomers and QDs, thus shortening the duration and avoiding the decomposition of thiol ligands (Scheme 3). Luo et al. demonstrated that N2H4 not only promoted the growth of 1-thioglycerol-stabilized CdTe QDs, but also enhanced the photoluminescence quantum yields (PLQYs) of the resulting QDs by avoiding the mercapto-ligand decomposition and sulfur embedment [126].

|
Download:
|
| Scheme 3. Schematic illustration of the electric double-layer of MPA-stabilized CdTe NCs. The presence of simple amines lowered the charges of Cd monomers. The adsorbed cation was predominantly Na+. Reprinted with permission [125]. Copyright 2010, Royal Society of Chemistry. | |
{kind=link}
The abovementioned aqueous anionic chalcogen precursors are either in the form of H2Se and H2Te gases, or in the form of NaHSe and NaHTe solutions, which require manipulation under isolation air. Due to the higher reactivity of selenium than tellurium, specific air-stable compounds, which will release selenium ions, should be more appropriate for the aqueous Se precursor. In 2000, Rogach et al. employed N, N-dimethylselenourea as the Se precursor to synthesize CdSe QDs with the assistance of microwave, which provided uniform heating [127]. With N, N-dimethylselenourea as the Se precursor, Chang's group also presented a simple photoassisted route to CdSe/CdS core-shell QDs [128]. PLQY of the coreshell CdSe/CdS QDs were improved to 60%. Zhu's group refluxed selenium powder in Na2SO3 aqueous solution and obtained a Na2SeSO3 solution [129]. The resulting Na2SeSO3 was employed as the Se precursor and further yielded a series of MSQDs at roomtemperature under ambient pressure, including CdSe, ZnSe, PbSe, Ag2Se, HgSe and Cu2-xSe. Note that the Na2SeSO3 should be stored in dark under inert conditions. Kalasad et al. found that the combination of N2H4 and selenium could form a soluble and airstable amine-Se complex [130]. In this dissolution process, N2H4 played the role of reducing agent and complexing agent. The amine-Se complex was further used to prepare high-quality aqueous CdTe QDs with PLQYs over 40%.
3.2. Cationic chalcogen precursorThe cationic chalcogen precursors are mainly in the form of SeO32- and TeO32- which are soluble in water. Compared with elemental selenium or tellurium, these chalcogenites are much cheaper and more stable. In 2003, Yang et al. synthesized CdSe and CdTe QDs at room-temperature, by using Na2SeO3 and K2TeO3 as the chalcogen precursors under γ-irradiation [131]. When irradiated by γ-ray, these chalcogenites are gradually reduced by reductive organic radicals and slowly release Se2- or Te2- ions. Resulted Se2- or Te2- ions combine with [Cd(NH3)4]2+ and further crystallize into CdSe and CdTe QDs. Although the reaction conditions are moderate and energy-saving, avoidless aggregation of the QDs and introduction of chalcogen dioxides are serious drawbacks of this protocol. In 2006, Dong's group developed a onepot aqueous synthesis method for CdTe QDs by using TeO32- as the Te precursor [132]. Experimentally, certain amount of CdCl2, NaBH4, trisodium citrate dehydrate and L-cysteine were simply mixed in water, followed by open-air reflux. The citrate was used to prevent the deposition of CdTeO3 before the formation of CdTe QDs. Later, Zou's group employed Na2SeO3 and Na2TeO3 as the chalcogen precursor, and analogously synthesized type-Ⅱ CdTe/ CdSe core/shell QDs via a two-step approach [133]. The emission of the obtained CdTe/CdSe core/shell QDs was highly bright and could be tuned from green to red by adjusting the thickness the of CdSe shell.
Apart from NaBH4, N2H4 is also used as reducing agent for cationic chalcogen precursor in aqueous synthesis. Li's group employed N2H4 to reduce Na2SeO3 at the water-oil interface and prepared CdSe, ZnSe, and ZnxCd1-xSe QDs [134]. Due to the weak reducibility of N2H4, the reaction should be operated in an autoclave at 160 ℃. The weak reducibility of N2H4 also leads to the failure in extending this method for the preparation of MTQDs. In this context, Zhou et al. introduced a stronger reducing agent, NaBH4, into the N2H4-promoted system and demonstrated a facile approach for room-temperature synthesis of highly emissive CdTe QDs [135]. Without pre-preparation for precursor, pH adjustment, inert atmosphere, and even energy consumption, CdTe QDs were obtained by just adding water, CdCl2, ligands, Na2TeO3, NaBH4 and N2H4 stepwise. The procedure is illustrated in Fig. 5. The addition sequence of the raw materials is validated to be very crucial, since the addition of Na2TeO3 prior to Cd source and thiol ligands may lead to the hydrolysis of Na2TeO3, and addition of N2H4 prior to NaBH4 may lead to incompletely reduced Te0 rather than Te2-. Emissions of the resulting QDs can be tuned by simply adjusting the feed ratio between the raw materials. This approach is also suitable for the synthesis of CdTe QDs with special capping ligands, such as 4-mercaptobenzoic acid, per-6-thio-α-cyclodextrin, and per-7-thio-β-cyclodextrin, which is hardly possible in conventional reflux method. Noteworthily, Zhou et al. also employed N2H4 to initialize the 1D self-assembly of the resulting CdTe QDs and act as monomers to polymerize with QDs capped by mercaptocarboxylic acids [136]. The resulted 1D self-assembly materials exhibit extremely high luminescence and excellent stability toward temperature, solvent and light due to the cross-linked structures, which are far superior to the CdTe QDs from conventional reflux methods. Emission of the 1D materials can be directly tuned by varying the size of the composed CdTe QD (Fig. 6). Although hydrazine is toxic and the amount of hydrazine is excessive, it can be easily recycled, thus reducing the impacts on environment. Overall, the H2H4-promoted method is facile and versatile, and holds great potential for industrial preparation of CdTe QDs-based phosphors.

|
Download:
|
| Fig. 5. Photographic illustration of the procedure for synthesizing CdTe QDs with red emission. Reprinted with permission [135]. Copyright 2011, American Chemical Society. | |
{kind=link}

|
Download:
|
| Fig. 6. PL images of the self-assembly materials composed of CdTe NCs with green (a), yellow (b), and red (c) emission. (d) The emission spectra of the self-assembly materials with different emission colors. Reprinted with permission [136]. Copyright 2014, American Chemical Society. | |
{kind=link}
4. Conclusions and outlook
In conclusion, we have summarized the recent green chemical synthesis routes to MSQDs and MTQDs. The synthetic chemistry might be considered variants of two systems: the organic phase system and the aqueous phase system. On the one hand, in the organic phase system, where QDs with good monodispersity and high crystallinity are usually yielded, conventional air-sensitive and toxic organophosphorus chalcogen precursors are basically replaced. The newly developed chalcogen precursors are not only phosphine-free, but also straightforward and highly reactive, thus reducing the requirement for dedicated equipments and lowering the energy consumption to a great extent. On the other hand, green synthetic methods in the aqueous phase system are not only featured for environmentally benign medium, but also greatly developed by designing and utilizing more stable and accessible chalcogen precursors to replace conventional ones based on hydrogen chalcogenide gases. It is important to stress that a hydrazine-promoted method in aqueous solution can produce high-quality CdTe QDs at room-temperature, spontaneously, which tremendously simplifies the operation process.
Despite the recent impressive progress in green synthetic methods for MSQDs and MTQDs, there is still large room for improvement. First of all, although water is deemed the ideal medium for green synthesis, the QDs yielded from the aqueous phase system are still less developed in types and less qualified in properties than those from the organic phase system. Further efforts should be devoted to the fundamental mechanisms in the aqueous phase system along with new synthetic methodologies to improve the performance of resulting products. Secondly, the current mature and prominent chalcogenide QDs are mainly heavy-metal-dependent, such as Cd, Pb, Hg and so forth. Taking into account the principles of green chemistry, alternatives containing metal elements with less toxicity and lower price are undoubtedly an important direction to pay attention to. Thirdly, in both the organic phase system and the aqueous phase system, the raw materials cannot be totally converted to high-quality QDs, because excess reagent precursors or capping ligands are generally applied to achieve composition or shape tuning. The atomic efficiency is a vital point for green synthesis that have been neglected. New approaches are desired to take full advantage of the raw materials and facilely recycle and reuse the excessive ones. Finally, although the current state-of-the-art green methods for MSQDs and MTQDs show great potentials for mass production, it is still challenging to transit these green technologies from laboratory to industrial production line. In short, we are expecting new advances in the green chemical methods, thus further extending their practical applications of MSQDs and MTQDs in the upcoming years.
AcknowledgmentsThis work was supported by the National Key research and Development Program of China (No. 2016YFB0401701), the 973 Program of China (No. 2014CB643503), the National Natural Science Foundation of China (NSFC, Nos. 21773088, 51425303), JLU Science and Technology Innovative Research Team (No. 2017TD- 06), National Postdoctoral Program for Innovative Talents (No. BX201700099), the China Postdoctoral Science Foundation (No. 2017M621207), and the Special Project from Ministry of Science and Technology of the People's Republic of China.
| [1] |
A.P. Alivisatos, Science 271 (1996) 933-937. DOI:10.1126/science.271.5251.933 |
| [2] |
D. Yu, F. Cao, Y. Gao, Y. Xiong, H. Zeng, Adv. Funct. Mater. 28 (2018) 1800248. DOI:10.1002/adfm.v28.19 |
| [3] |
J. Song, J. Li, X. Li, et al., Adv. Mater. 27 (2015) 7162-7167. DOI:10.1002/adma.201502567 |
| [4] |
H. Ding, S. Yu, H. Xiong, ACS Nano 10 (2016) 481-494. |
| [5] |
F. Qu, Y. Yuan, M. Yang, Chem. Mater. 29 (2017) 969-974. DOI:10.1021/acs.chemmater.6b03435 |
| [6] |
Z. He, Z. Jin, M. Zhan, et al., Chin. Chem. Lett. 28 (2017) 1851-1856. DOI:10.1016/j.cclet.2017.07.012 |
| [7] |
Y. Li, C. Xu, C. Shu, X. Hou, P. Wu, Chin. Chem. Lett. 28 (2017) 1961-1964. DOI:10.1016/j.cclet.2017.04.027 |
| [8] |
L. Sorensen, G.F. Strouse, A.E. Stiegman, Adv. Mater. 18 (2006) 1965-1967. |
| [9] |
Z. Li, W. Yao, L. Kong, Y. Zhao, L. Li, J. Am. Chem. Soc. 138 (2015) 12430-12433. |
| [10] |
S. Huang, Z. Li, L. Kong, et al., J. Am. Chem. Soc. 137 (2016) 5749-5752. |
| [11] |
B. Chen, N. Pradhan, H. Zhong, J. Phys. Chem. Lett. 9 (2018) 435-445. DOI:10.1021/acs.jpclett.7b03037 |
| [12] |
W. Li, R. Zamani, P. Rivera Gil, et al., J. Am. Chem. Soc. 135 (2013) 7098-7101. DOI:10.1021/ja401428e |
| [13] |
V.B. Llorente, V.M. Dzhagan, N. Gaponik, et al., J. Phys. Chem. C 121 (2017) 18244-18253. DOI:10.1021/acs.jpcc.7b05334 |
| [14] |
C. Coughlan, M. Iba'ñez, O. Dobrozhan, et al., Chem. Rev. 117 (2017) 5865-6109. DOI:10.1021/acs.chemrev.6b00376 |
| [15] |
J. Zheng, B. Dai, J. Liu, et al., ACS Appl. Mater. Interfaces 8 (2016) 35426-35434. DOI:10.1021/acsami.6b11058 |
| [16] |
A. Agrawal, S.H. Cho, O. Zandi, et al., Chem. Rev. 118 (2018) 3121-3207. DOI:10.1021/acs.chemrev.7b00613 |
| [17] |
T.C. Harman, P.J. Taylor, M.P. Walsh, B.E. LaForge, Science 297 (2002) 2229-2232. DOI:10.1126/science.1072886 |
| [18] |
M. Iba'ñez, R. Zamani, S. Gorsse, et al., ACS Nano 7 (2013) 2573-2586. DOI:10.1021/nn305971v |
| [19] |
J. Zhou, R. Yang, J. Appl. Phys. 110 (2011) 084317. DOI:10.1063/1.3653263 |
| [20] |
T.C. Harman, P.J. Taylor, D.L. Spears, M.P. Walsh, J. Electron. Mater. 29 (2000) L1-L2. DOI:10.1007/s11664-000-0117-1 |
| [21] |
M.C. Kovalenko, W. Heiss, E.V. Shevchenko, et al., J. Am. Chem. Soc. 129 (2007) 11354-11355. DOI:10.1021/ja074481z |
| [22] |
A. Aboulaich, L. Balan, J. Ghanbaja, et al., Chem. Mater. 23 (2011) 3706-3713. DOI:10.1021/cm2012928 |
| [23] |
C. Wang, S. Xu, Y. Wang, Z. Wang, Y. Cui, J. Mater. Chem. C 2 (2014) 660-666. DOI:10.1039/C3TC31602E |
| [24] |
C. Wang, J. Xu, Y. Wang, et al., Adv. Funct. Mater. 26 (2016) 4274-4282. DOI:10.1002/adfm.v26.24 |
| [25] |
P. Wu, X. Yan, Chem. Soc. Rev. 42 (2013) 5489-5521. DOI:10.1039/c3cs60017c |
| [26] |
B. Zhao, Y. Yao, K. Yang, et al., Nanoscale 6 (2014) 12345-12349. DOI:10.1039/C4NR03490B |
| [27] |
C. Wang, X. Gao, Q. Ma, X. Su, J. Mater. Chem. 19 (2009) 7016-7022. DOI:10.1039/b909546b |
| [28] |
X. Gao, G. Tang, X. Su, Biosens. Bioelectron. 36 (2012) 75-80. DOI:10.1016/j.bios.2012.03.042 |
| [29] |
Q. Peng, Y. Dong, Z. Deng, Y. Li, Inorg. Chem. 41 (2002) 5249-5254. DOI:10.1021/ic0257266 |
| [30] |
X. Ren, L. Pang, Y. Zhang, et al., J. Mater. Chem. A 3 (2015) 10693-10697. DOI:10.1039/C5TA02198G |
| [31] |
C. Wadia, Y. Wu, S. Gul, et al., Chem. Mater. 21 (2009) 2568-2570. DOI:10.1021/cm901273v |
| [32] |
J. Wang, H. Han, J. Colloid Interface Sci. 351 (2010) 83-87. DOI:10.1016/j.jcis.2010.07.025 |
| [33] |
M. Li, Y. Ge, Q. Chen, et al., Talanta 72 (2007) 89-94. DOI:10.1016/j.talanta.2006.09.028 |
| [34] |
H. Li, C. Han, Chem. Mater. 20 (2008) 6053-6059. DOI:10.1021/cm8009176 |
| [35] |
H. Xiong, D.G. Shchukin, H. Möhwald, Y. Xu, Y. Xia, Angew. Chem. Int. Ed. 48 (2009) 2727-2731. DOI:10.1002/anie.200805590 |
| [36] |
J. Zhu, Y. Koltypin, A. Gedanken, Chem. Mater. 12 (2000) 73-78. DOI:10.1021/cm990380r |
| [37] |
M.J. Murcia, D.L. Shaw, H. Woodruff, et al., Chem. Mater. 18 (2006) 2219-2225. DOI:10.1021/cm0505547 |
| [38] |
Q. Shen, L. Jiang, J. Miao, W. Hou, J.J. Zhu, Chem. Commun. 14 (2008) 1683-1685. |
| [39] |
S. Gorer, J.A. Ganske, J.C. Hemminger, R.M. Penner, J. Am. Chem. Soc. 120 (1998) 9584-9593. DOI:10.1021/ja981676l |
| [40] |
S. Li, H. Zhao, D. Tian, Mat. Sci. Semicon. Proc. 16 (2013) 149-153. DOI:10.1016/j.mssp.2012.05.013 |
| [41] |
D. Gopalakrishnan, D. Damien, B. Li, et al., Chem. Commun. 51 (2015) 6293-6296. DOI:10.1039/C4CC09826A |
| [42] |
D.V. Freitas, J.M.M. Dias, S.G.B. Passos, et al., Green Chem. 16 (2014) 3247-3254. DOI:10.1039/c4gc00300d |
| [43] |
S.R. Sturzenbaum, M. Hockner, A. Panneerselvam, et al., Nat. Nanotech. 8 (2013) 57-60. DOI:10.1038/nnano.2012.232 |
| [44] |
H. Bao, N. Hao, Y. Yang, D. Zhao, Nano Res. 3 (2010) 481-489. DOI:10.1007/s12274-010-0008-6 |
| [45] |
H. Bao, Z. Lu, X. Cui, et al., Acta Biomater. 6 (2010) 3534-3541. DOI:10.1016/j.actbio.2010.03.030 |
| [46] |
Q.Y. Luo, Y. Lin, Y. Li, et al., Small 10 (2014) 699-704. DOI:10.1002/smll.201301940 |
| [47] |
V. Kloper, R. Osovsky, J. Kolny-olesiak, A. Sashchiuk, E. Lifshiitz, J. Phys. Chem. C 111 (2007) 10336-11034. DOI:10.1021/jp0708906 |
| [48] |
S. Kim, B. Fisher, H. Eisler, M. Bawendi, J. Am. Chem. Soc. 125 (2003) 11466-11467. DOI:10.1021/ja0361749 |
| [49] |
J. Zhang, J. Wang, T. Yan, et al., J. Mater. Chem. B 5 (2017) 8152-8160. DOI:10.1039/C7TB02324C |
| [50] |
S. Marre, J. Park, J. Rempel, et al., Adv. Mater. 20 (2008) 4830-4834. DOI:10.1002/adma.v20:24 |
| [51] |
J. van Embden, A.S.R. Chesman, J.J. Jasieniak, Chem. Mater. 27 (2015) 2246-2285. DOI:10.1021/cm5028964 |
| [52] |
V. Lesnyak, N. Gaponik, A. Eychmüller, Chem. Soc. Rev. 42 (2013) 2905-2929. DOI:10.1039/C2CS35285K |
| [53] |
M.L. Steigerwald, E. Brus, Acc. Chem. Res. 23 (1990) 183-188. DOI:10.1021/ar00174a003 |
| [54] |
C.N.R. Rao, S.R.C. Vivekchand, K. Biswasa, A. Govindaraj, Dalton Trans. 34 (2007) 3728-3749. |
| [55] |
O. Masala, R. Seshadri, Annu. Rev. Mater. Res. 34 (2004) 41-81. DOI:10.1146/annurev.matsci.34.052803.090949 |
| [56] |
T. Trindade, Chem. Mater. 13 (2001) 3843-3858. DOI:10.1021/cm000843p |
| [57] |
D. Fan, M. Afzaal, M.A. Mallik, et al., Coordin. Chem. Rev. 251 (2007) 1878-1888. DOI:10.1016/j.ccr.2007.03.021 |
| [58] |
F. Wang, A. Dong, W.E. Buhro, Chem. Rev. 116 (2016) 10888-10933. DOI:10.1021/acs.chemrev.5b00701 |
| [59] |
X. Michalet, F.F. Pinaud, L.A. Bentolila, et al., Science 307 (2005) 538-544. DOI:10.1126/science.1104274 |
| [60] |
M.L. Steigerwald, A.P. Alivisatos, J.M. Gibson, et al., J. Am. Chem. Soc. 110 (1988) 3046-3050. DOI:10.1021/ja00218a008 |
| [61] |
C.B. Murray, D.J. Noms, M.G. Bawendi, J. Am. Chem. Soc. 115 (1993) 8706-8715. DOI:10.1021/ja00072a025 |
| [62] |
Z. Peng, X. Peng, J. Am. Chem. Soc. 123 (2001) 183-184. DOI:10.1021/ja003633m |
| [63] |
L.S. Li, H. Wang, Y. Liu, et al., J. Colloid Interface Sci. 308 (2007) 254-257. DOI:10.1016/j.jcis.2006.12.035 |
| [64] |
Y. Liu, Y. Tang, Y. Ning, et al., J. Mater. Chem. 20 (2010) 4451-4458. DOI:10.1039/c0jm00115e |
| [65] |
L. Qu, Z. Peng, X. Peng, Nano Lett. 1 (2001) 333-337. DOI:10.1021/nl0155532 |
| [66] |
Z. Pan, H. Zhang, K. Chen, et al., ACS Nano 6 (2012) 3982-3991. DOI:10.1021/nn300278z |
| [67] |
S. Keuleyan, E. Lhuillier, P. Guyot-Sionnest, J. Am. Chem. Soc. 133 (2011) 16422-164244. DOI:10.1021/ja2079509 |
| [68] |
Y. Pan, H. Bai, L. Pan, et al., J. Mater. Chem. 22 (2012) 23593-23601. |
| [69] |
J. Du, Z. Du, J. Hu, L. Wan, et al., J. Am. Chem. Soc. 138 (2016) 4201-4209. DOI:10.1021/jacs.6b00615 |
| [70] |
R. Gomes, A. Hassinen, A. Szczygiel, et al., J. Phys. Chem. Lett. 2 (2011) 145-152. DOI:10.1021/jz1016729 |
| [71] |
D. Deng, C. Hao, S. Sen, et al., J. Am. Chem. Soc. 139 (2017) 16630-16639. DOI:10.1021/jacs.7b07838 |
| [72] |
S. Deka, A. Genovese, Y. Zhang, et al., J. Am. Chem. Soc. 132 (2010) 8912-8914. DOI:10.1021/ja103223x |
| [73] |
M. Sun, X. Yang, J. Phys. Chem. C 113 (2009) 8701-8709. DOI:10.1021/jp811308h |
| [74] |
J. Li, H. Wang, L. Lin, Q. Fang, X. Peng, J. Am. Chem. Soc. 140 (2018) 5474-5484. DOI:10.1021/jacs.8b01296 |
| [75] |
K. Yu, X. Liu, T. Qi, et al., Nat. Commun. 7 (2016) 12223. DOI:10.1038/ncomms12223 |
| [76] |
M. Liu, K. Wang, L. Wang, et al., Nat. Commun. 8 (2017) 15467. DOI:10.1038/ncomms15467 |
| [77] |
T. Trindade, P. O'Brien, Adv. Mater. 8 (1996) 161-163. |
| [78] |
T. Trindade, P. O'Brien, Chem. Mater. 9 (1997) 523-530. DOI:10.1021/cm960363r |
| [79] |
M. Chunggaze, J. McAleese, P. O'Brien, D.J. Otway, Chem. Commun. 7 (1998) 833-834. |
| [80] |
B. Ludolph, M.A. Malik, P. O'Brien, N. Revaprasadu, Chem. Commun. 17 (1998) 1849-1850. |
| [81] |
W.S. Chang, Y.F. Lin, B. Sarkar, et al., Dalton Trans. 39 (2010) 2821-2830. DOI:10.1039/b921503d |
| [82] |
M.A. Malik, N. Revaprasadu, P. O'Brien, Chem. Mater. 13 (2001) 913-920. DOI:10.1021/cm0011662 |
| [83] |
J. Jasieniak, C. Bullen, J. van Embden, P. Mulvaney, J. Phys. Chem. B 109 (2005) 20665-20668. DOI:10.1021/jp054289o |
| [84] |
Z. Deng, L. Cao, F. Tang, B. Zou, J. Phys. Chem. B 109 (2005) 16671-16675. DOI:10.1021/jp052484x |
| [85] |
S. Sapra, A.L. Rogach, J. Feldmann, J. Mater. Chem. 16 (2006) 3391-3395. DOI:10.1039/b607022a |
| [86] |
C. Wang, Y. Jiang, G. Li, et al., J. Cryst. Growth 310 (2008) 2890-2894. DOI:10.1016/j.jcrysgro.2008.01.059 |
| [87] |
X. Liu, Y. Jiang, C. Wang, et al., J. Cryst. Growth 312 (2010) 2656-2660. DOI:10.1016/j.jcrysgro.2010.06.009 |
| [88] |
B. Wang, Y. Jiang, C. Liu, et al., Phys. Status Solidi A 209 (2012) 306-312. DOI:10.1002/pssa.201127552 |
| [89] |
S. Flamee, M. Cirillo, S. Abe, et al., Chem. Mater. 25 (2013) 2476-2483. DOI:10.1021/cm400799e |
| [90] |
C. Pu, J. Zhou, R. Lai, et al., Nano Res. 6 (2013) 652-670. DOI:10.1007/s12274-013-0341-7 |
| [91] |
Y.A. Yang, H. Wu, K.R. Williams, Y.C. Cao, Angew. Chem. Int. Ed. 44 (2005) 6712-6715. |
| [92] |
O. Chen, X. Chen, Y. Yang, et al., Angew. Chem. Int. Ed. 47 (2008) 8638-8641. DOI:10.1002/anie.v47:45 |
| [93] |
K. Yu, J. Ouyang, B. Md.Zaman, et al., J. Phys. Chem. C 113 (2009) 3390-3401. DOI:10.1021/jp809990a |
| [94] |
J. Ouyang, M. Vincent, D. Kingston, et al., J. Phys. Chem. C 113 (2009) 5193-5200. DOI:10.1021/jp8110138 |
| [95] |
R. Wang, O. Calvignanello, C.I. Ratcliffe, et al., J. Phys. Chem. C 113 (2009) 3402-3408. DOI:10.1021/jp810325z |
| [96] |
J. Ouyang, B. Md.Zaman, F.J. Yan, et al., J. Phys. Chem. C 112 (2008) 13805-13811. DOI:10.1021/jp803845n |
| [97] |
Y. Wei, J. Yang, A.W.H. Lin, J.Y. Ying, Chem. Mater. 22 (2010) 5672-5677. DOI:10.1021/cm101308f |
| [98] |
Y. Liu, D. Yao, L. Shen, et al., J. Am. Chem. Soc. 134 (2012) 7207-7210. DOI:10.1021/ja300064t |
| [99] |
D. Yao, H. Liu, Y. Liu, et al., Nanoscale 7 (2015) 18570-18578. DOI:10.1039/C5NR04856G |
| [100] |
J. Zhang, K. Sun, A. Kumbhar, J. Fang, J. Phys. Chem. C 112 (2008) 5454-5458. DOI:10.1021/jp711778u |
| [101] |
J. Zhang, S. Jin, H.C. Fry, et al., J. Am. Chem. Soc. 133 (2011) 15324-15327. DOI:10.1021/ja206309h |
| [102] |
H. Shen, H. Wang, X. Chen, et al., Chem. Mater. 22 (2010) 4756-4761. DOI:10.1021/cm1013009 |
| [103] |
K. Ramasamy, D. Mazumdar, R.D. Bennett, A. Gupta, Chem. Commun. 48 (2012) 5656-5658. DOI:10.1039/c2cc32021e |
| [104] |
D. Yao, Y. Liu, W. Zhao, et al., Nanoscale 5 (2013) 9593-9597. DOI:10.1039/c3nr03553k |
| [105] |
D. Yao, W. Xin, Z. Liu, et al., ACS Appl. Mater. Interfaces 9 (2017) 9840-9848. DOI:10.1021/acsami.6b16407 |
| [106] |
J.A. Dahl, B.L.S. Maddux, J.E. Hutchison, Chem. Rev. 107 (2007) 2228-2269. DOI:10.1021/cr050943k |
| [107] |
X. Peng, Chem.-Eur. J. 8 (2002) 334-339. |
| [108] |
U. Resch, H. Weller, A. Henglein, Langmuir 5 (1989) 1015-1020. DOI:10.1021/la00088a023 |
| [109] |
T. Rajh, O.I. Mi ci c, A.J. Nozik, J. Phys. Chem. 97 (1993) 11999-12003. DOI:10.1021/j100148a026 |
| [110] |
A.L. Rogach, A. Kornowski, M. Gao, A. Eychmüller, H. Weller, J. Phys. Chem. B 103 (1999) 3065-3069. DOI:10.1021/jp984833b |
| [111] |
A. Shavel, N. Gaponik, A. Eychmüller, J. Phys. Chem. B 108 (2004) 5905-5908. DOI:10.1021/jp037941t |
| [112] |
N. Gaponik, D.V. Talapin, A.L. Rogach, K. Hoppe, et al., J. Phys. Chem. B 106 (2002) 7177-7185. |
| [113] |
A. Rogach, S. Kershaw, M. Burt, et al., Adv. Mater. 11 (1999) 552-555. |
| [114] |
Y. Tian, T. Newton, N.A. Kotov, D.M. Guldi, J.H. Fendler, J. Phys. Chem. 100 (1996) 8927-8939. DOI:10.1021/jp951965l |
| [115] |
J.M. Lalancette, M. Arnac, Can. J. Chem. 47 (1969) 3695-3697. DOI:10.1139/v69-611 |
| [116] |
E. Hao, H. Zhang, B. Yang, H. Ren, J. Shen, J. Colloid Interface Sci. 238 (2001) 285-290. DOI:10.1006/jcis.2001.7472 |
| [117] |
H. Zhang, Z. Zhou, B. Yang, M. Gao, J. Phys. Chem. B 107 (2003) 8-13. DOI:10.1021/jp025910c |
| [118] |
D. Zhou, J. Han, Y. Liu, et al., J. Phys. Chem. C 114 (2010) 22487-22492. DOI:10.1021/jp108708n |
| [119] |
H. Zhang, Z. Cui, Y. Wang, et al., Adv. Mater. 15 (2003) 777-780. DOI:10.1002/adma.200304521 |
| [120] |
H. Zhang, C. Wang, M. Li, et al., Adv. Mater. 17 (2005) 853-857. |
| [121] |
H. Zhang, D. Wang, B. Yang, H. Möhwald, J. Am. Chem. Soc. 128 (2006) 10171-10180. DOI:10.1021/ja061787h |
| [122] |
H. Zhang, D. Wang, H. Möhwald, Angew. Chem. Int. Ed. 45 (2006) 748-751. |
| [123] |
H. Zhang, Y. Liu, J. Zhang, et al., J. Phys. Chem. C 112 (2008) 1885-1889. |
| [124] |
H. Zhang, Y. Liu, C. Wang, et al., Chemphyschem 9 (2008) 1309-1316. |
| [125] |
J. Han, H. Zhang, H. Sun, D. Zhou, B. Yang, Phys. Chem. Chem. Phys. 12 (2010) 332-336. DOI:10.1039/B919655B |
| [126] |
X. Luo, J. Han, Y. Ning, et al., J. Mater. Chem. 21 (2011) 6569-6575. DOI:10.1039/c0jm04425c |
| [127] |
A.L. Rogach, D. Nagesha, J.W. Ostrander, M. Giersig, N.A. Kotov, Chem. Mater. 12 (2000) 2676-2685. DOI:10.1021/cm000244i |
| [128] |
Y. Lin, M. Hsieh, C. Liu, H. Chang, Langmuir 21 (2005) 728-734. DOI:10.1021/la049489q |
| [129] |
X. Ma, X. Qian, J. Yin, Z. Zhu, J. Mater. Chem. 12 (2002) 663-666. DOI:10.1039/b107173b |
| [130] |
M.N. Kalasad, M.K. Rabinal, B.G. Mulimani, Langmuir 25 (2009) 12729-12735. DOI:10.1021/la901798y |
| [131] |
Q. Yang, K. Tang, F. Wang, C. Wang, Y. Qian, Mater. Lett. 57 (2003) 3508-3512. DOI:10.1016/S0167-577X(03)00117-4 |
| [132] |
H. Bao, E. Wang, S. Dong, Small 2 (2006) 476-480. |
| [133] |
R. Zeng, T. Zhang, J. Liu, et al., Nanotechnology 20 (2009) 095102. DOI:10.1088/0957-4484/20/9/095102 |
| [134] |
J. Ge, S. Xu, J. Zhuang, et al., Inorg. Chem. 45 (2006) 4922-4927. DOI:10.1021/ic051598k |
| [135] |
D. Zhou, M. Lin, Z. Chen, et al., Chem. Mater. 23 (2011) 4857-4862. DOI:10.1021/cm202368w |
| [136] |
D. Zhou, M. Liu, M. Lin, et al., ACS Nano 8 (2014) 10569-10581. DOI:10.1021/nn5040444 |