 2019, Vol. 30
2019, Vol. 30 


b State Key Laboratory of Bioorganic and Natural Products Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
The emergence and evolution of click chemistry have greatly facilitated the development of the fields of chemical biology and material science [1]. The copper catalyzed azide–alkyne cycloaddition (CuAAC)pioneered by Sharpless and co-workers [2] remainsoneof the most powerful tools of clickchemistry, despitenumerous endeavors to develop various click reactions [1c, d, g]. CuAAC provides an expedient and modular approach to prepare small molecule probes of biological interest; biotin, photoactivatable groups, and fluorophores can be readily installed onto the small molecule core at a late stage [1b]. In recent years, sequential click reactions have received increasing attention [3]. The programmed reaction sequence allows rapid construction of multifunctional small molecule probes and polymers. For instance, an affinity-based bifunctional probe for cellular target identification usually contain a photoactivatable group (such as a diazirine) for crosslinking with the target protein and a biotin tag for pull-down [4], and it is more practical to introduce the photoactivatable group by a click reaction at a late stage rather than to carry on the fairly labile functionality through a de novo synthesis. In this case, sequential click reactions are highly desired. Although one can envision a variety of combinations of click reactions, the most attractive and straightforward strategy of using double CuAAC reactions to install different functionalities is hampered by the narrow window for differentiation of the substrate reactivity. Recently, we reported the LiCl mediated elimination of alkenyl triflates for the preparation of alkynes under mild conditions [5], which permits a strategy of masking terminal alkynes as alkenyl triflates in a click process. Thus, we envision a programmed sequence of two copper mediated cycloaddition reactions starting from a substrate containing a terminal alkyne and an alkenyl triflate which can be easily converted into an terminal alkyne. The first click event should occur under standard CuAAC conditions, while the second step requires an efficientone-potprotocolon thebasis ofour previous discovery [5]. Herein, we describe such a protocol for copper-mediated azide–alkyne cycloaddition using alkenyl triflate precursors.
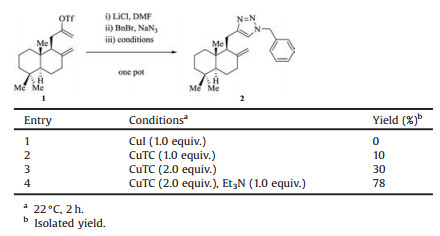
We investigated the conditions for the one-pot elimination/azide formation/cycloaddition sequence, using readily available alkenyl triflate 1 [5] as a precursor. The results were summarized in Table 1. In the presence of LiCl, elimination of 1 proceeded in DMF at ambient temperature to give the desired terminal alkyne [5]. Benzyl azidewas then generated in situ from NaN3 and BnBr through an SN2 reaction; DMF also served as a good solvent for the substitution. Thus, we needed a suitable copper salt or complex as a promoter for the cycloaddition, which had to be compatible to the reaction system. The commonly used CuI [6] proved to be ineffective under these conditions (entry 1). Interestingly, copper(I) thiophene-2- carboxylate (CuTC) [5, 7] led toformation of a small amount of 1, 2, 3- triazole 2 (entry 2). Increasing the equivalents of CuTC improved the yield of 2 to 30%, which was still unsatisfactory from a synthetic perspective. To our delight, Et3N was found to be a crucial additive for this one-pot process, and 2 was obtained in 78% isolated yield from 1. Et3N presumably buffered the acid generated through the elimination process and might promote the formation of the copper acetylide intermediate as well. Of note, the amine can be added before the SN2 reaction; this order change had no bearing on the overall efficiency.
|
|
Table 1 Studies of the conditions for the one-pot triflate elimination/azide formation/ cycloaddition sequence |
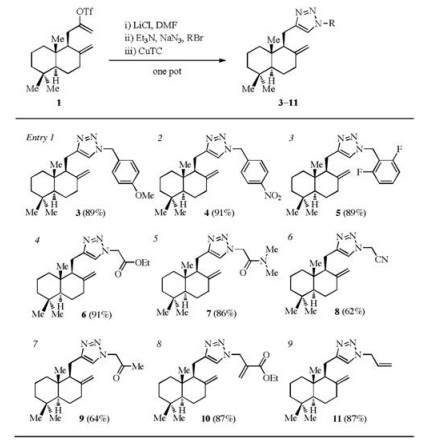
Having established the optimal conditions, we examined a series of azides (generated in situ from NaN3 and corresponding alkyl bromides) for the one-pot process, as shown in Table 2. 1, 2, 3- Triazoles 3–11 were obtained in good to excellent yields (entries 1– 9). Substituted benzyl azides with electron-rich (entry 1) or electron-deficient (entries 2 and 3) aromatic rings all performed well under the standard conditions. α-Azido carbonyl compounds (entries 4–7) were suitable substrates as well, which would allow facile incorporation of a photoactivatable group or a biotin tag. To our delight, the reactions of allyl azides (entries 8 and 9) proceeded with good efficiency, which may offer an alternative type of linkers of improved stability compared with esters and amides.
|
|
Table 2 Scope of the azides (generated in situ) suitable for the one-pot sequence |
A general procedure for the one-pot sequence is described below. To a stirred solution of alkenyl triflate 1 (34.2 mg, 0.090 mmol) in DMF (300 μL) was added anhydrous LiCl (15.3 mg, 0.361 mmol) at 22 ℃. The mixture was allowed to stir at that temperature for 1.5 h before Et3N (9.1 mg, 12.5 μL, 0.090 mmol), NaN3 (8.8 mg, 0.135 mmol), and the bromide (0.135 mmol) were sequentially added. The resultant mixture was stirred at 22 ℃ for 2.5 h, and CuTC (34.3 mg, 0.180 mmol) was added. The mixture was allowed to stir at that temperature for 2 h before it was quenched with saturated aq. NaHCO3 (2 mL) and extracted with EtOAc (5 × 2 mL). The combined organic phases were washed with brine (5 mL), dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The residue was subjected to preparative thin layer chromatography for purification using EtOAc/petroleum ether (1:3) as eluent to give the 1, 2, 3-triazole. Characterization of compounds 2–11 is included in the Supporting information.
In summary, we have developed an efficient protocol for copper-mediated azide–alkyne cycloaddition using alkenyl triflate precursors. Three sequential reactions, namely the LiCl-promoted elimination for alkyne formation, the nucleophilic substitution for azide formation, and the cycloaddition for the triazole formation, proceeded smoothly in one pot. This protocol may find further use in the construction of bifunctional small molecule probes for target identification, imaging, and other chemical biology studies.
AcknowledgmentsWe thank Xiaowen Yang and Dr. Wenhao Zhang for helpful discussions. Financial support was provided by the National Natural Science Foundation of China (Nos. 21525209, 21621002, 21772225, and 21761142003), the Chinese Academy of Sciences (Strategic Priority Research Program (No. XDB20000000) and Key Research Program of Frontier Sciences (No. QYZDB-SSW-SLH040)), Shanghai Science and Technology Commission (Nos. 15JC1400400 and 17XD1404600), the National Program for Support of TopNotch Young Professionals of China, and the K. C. Wong Education Foundation.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.06.001.
| [1] |
(a) H.C. Kolb, M.G. Finn, K.B. Sharpless, Angew. Chem. Int. Ed. 40 (2001) 2004-2021; (b) H.C. Kolb, K.B. Sharpless, Drug Discov. Today 8 (2003) 1128-1137; (c) J. Dong, L. Krasnova, M.G. Finn, K.B. Sharpless, Angew. Chem. Int. Ed. 53 (2014) 9430-9448; (d) J.C. Jewett, C.R. Bertozzi, Chem. Soc. Rev. 39 (2010) 1272-1279; (e) P.L. Golas, K. Matyjaszewski, Chem. Soc. Rev. 39 (2010) 1338-1354; (f) J.E. Moses, A.D. Moorhouse, Chem. Soc. Rev. 36 (2007) 1249-1262; (g) W. Xi, T.F. Scott, C.J. Kloxin, C.N. Bowman, Adv. Funct. Mater. 24 (2014) 2572-2590. |
| [2] |
V.V. Rostovtsev, L.G. Green, V.V. Fokin, K.B. Sharpless, Angew. Chem. Int. Ed. 41 (2002) 2596-2599. DOI:10.1002/(ISSN)1521-3773 |
| [3] |
(a) N. Münster, P. Nikodemiak, U. Koert, Org. Lett. 18 (2016) 4296-4299; (b) R.R. Ramsubhag, G.B. Dudley, Org. Biomol. Chem. 14 (2016) 5028-5031; (c) M.Z.C. Hatit, C.P. Seath, A.J.B. Watson, G.A. Burley, J. Org. Chem. 82 (2017) 5461-5468. |
| [4] |
S. Pan, H. Zhang, C. Wang, S.C.L. Yao, S.Q. Yao, Nat. Prod. Rep. 33 (2016) 612-620. DOI:10.1039/C5NP00101C |
| [5] |
X. Yang, D. Wu, Z. Lu, H. Sun, A. Li, Org. Biomol. Chem. 14 (2016) 5591-5594. DOI:10.1039/C6OB00345A |
| [6] |
L.Y. Wu, Y.X. Xie, Z.S. Chen, Y.N. Niu, Y.M. Liang, Synlett (2009) 1453-1456. |
| [7] |
J. Raushel, V.V. Fokin, Org. Lett. 12 (2010) 4952-4955. DOI:10.1021/ol102087r |