 2019, Vol. 30
2019, Vol. 30 


b School of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou 225002, China
Dialkylideneacetones serve as the key precursors to construct/ synthesize many useful organic skeletons or natural products, such as pyrimidines, 2, 7-disubstituted tropones, cystodytins, etc. [1-3]. They are also widely employed as bioactive components in antiangiogenic reagent, quinine reductase inducer and arginine methyltransferase inhibitor in biochemistry studies [4-7], as agrochemical, pharmaceutical and perfume intermediates and ligands in fine chemical industry [8-11], and as liquid crystalline polymer units in materials science [12]. Like simple α, β-unsaturated ketones, dialkylideneacetones could be prepared through the Claisen-Schmidt reactions of acetone with aldehydes for the high atom utilization efficiency form the industrial viewpoint [13, 14]. However, the present methods were catalyzed/promoted by strong acids or bases [15-19], Lewis acid catalysts such as Cu(OTf)2 [20], FeCl3 [21], TiCl3(OTf) [22], SmI3 [23], Yb(OTf)3 [24], etc., noble metal catalysts such as RuCl3 [25], TMSCl/Pd-C [26], or supported catalysts/dehydrants such as KF-Al2O3 [27], P2O5/SiO2 [28], etc., which were corrosive, expensive, or required tedious fabrication methods or high catalyst loadings. Moreover, although there are many reports on symmetrically substituted dialkylideneacetones, method for the synthesis of dialkylideneacetones with different alkylidenes has not been well documented yet.
Our group aims to develop the green synthetic methods with industrial application potential [29-34]. Recently, it was found that Ca(OH)2, the cheap, mild and common but rarely used inorganic base, could serve as the efficient catalyst in dialkylideneacetone synthesis. The method required very low catalyst loading (10 mol%) and could use dilute alcohol as the green solvent (20% aqueous solution, v/v). It could be expanded to at least 100 mmol scales to produce the related dialkylideneacetones in gram scale. Herein, we wish to report the details of the method.
The Ca(OH)2-catalyzed reaction of (E)-4-phenylbut-3-en-2-one (1a) with benzaldehyde (2a) were initially tested. After heating 1 mmol of 1a with equivalent 2a in 1 mL of EtOH in the presence of 0.2 mmol of Ca(OH)2 at 60 ℃ for 48 h, the desired product 3a could be isolated in 47% yield by preparative thin layer chromatography Scheme 1). In the process, partial of the Ca(OH)2 was insoluble and precipitated at the bottom of the reaction tube. Therefore, water was then introduced into the solvent to enhance the Ca(OH)2 solubility. The product yield was enhanced with elevated H2O content and 20% aqueous EtOH (volume concentration) was found to be the most preferable solvent, affording 3a in 80% yield (Fig. 1). No reaction occurred in the absence of EtOH, although Ca(OH)2 dissolved completely in the case. This was probably due to the low solubility of the organic reactants in water, as after the addition of 2 mol% of sodium dodecyl sulfate or benzyldodecyldimethylammonium bromide as surfactant, the same reactions in water could afford the desired product 3a in 87% and 39% yields, respectively. Thus, EtOH, as the water-soluble organic solvent, might facilitate the contact of the organic reactants with Ca(OH)2 to promote the reaction, and it was better than the sodium dodecyl sulfate surfactant for the solid waste-free process.

|
Download:
|
| Scheme 1. Synthesis of dialkylideneacetones catalyzed by Ca(OH)2 | |

|
Download:
|
| Fig. 1. Experimental results of the reactions performed in aqueous EtOH at different volume concentrations | |
Additional parallel experiments were conducted to optimize the reaction conditions. Elevated Ca(OH)2 amount could hardly improve the reaction, while the excess catalyst precipitated due to its low solubility (Table 1, entry 1). Considering the higher turnover number and the acceptable product yield, Ca(OH)2/1a = 10% was then screened out to be the better condition (Table 1, entries 3 vs. 1, 2, 4). The reaction was retarded at reduced temperature (Table 1, entry 5), and afforded 3a in the highest yield (83%) at 80 ℃ (Table 1, entries 7 vs. 6, 8).
|
|
Table 1 Condition optimizations for the reaction of (E)-4-phenylbut-3-en-2-one (1a) with benzaldehyde (2a). |

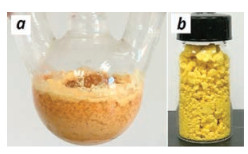
Under the optimized conditions, the reaction of 1a with benzaldehyde (2a) was magnified into 10 mmol scale and the desired product 3a could be obtained in 81% yield through column chromatography isolation (Table 2, entry 1). Further magnified reaction at 100 mmol was conducted and in the process, the product 3a precipitated and could be isolated by a simple filtration (Fig. 2a). After washing with water to remove the Ca(OH)2 catalyst and drying under infrared lamp irradiation, pure 3a was obtained as a yellow solid in 80% yield (Fig. 2b and Table 2, entry 2). 4- Methylbenzaldehyde (2b), the electron-enriched aldehyde, could also react with 1a to produce the related dialkylideneacetone 3b in good yield at 100 mmol scale (Table 2, entry 3), but for 4- methoxybenzaldehyde (2c), since a series of unidentified byproduct was generated, no precipitation was observed in the 100 mmol-scale reaction and the product was isolated in 51% yield by column chromatography in a 10 mmol-scale reaction (Table 2, entry 4). Notably, electron-deficient aldehydes, such as 2d–e, were preferable for the reaction, affording the desired products in excellent to almost quantitative yields (Table 2, entries 5 and 6). The reactions with heterocycle-substituted aldehydes 2f– g also led to excellent product yields (Table 2, entries 7 and 8). Aliphatic and alkenyl aldehydes 2h–i were also applicable to produce 3h–i in 32%–55% yields (Table 2, entries 9 and 10). Because Ca(OH)2 catalyst was used in very low loading (10 mol%) and might be deactivated by the carboxylic acid impurities, the aldehydes should be purified by distillation or recrystallization before using.
|
|
Table 2 Reactions of (E)-4-phenylbut-3-en-2-one (1a) with aldehyde (2) at magnified reaction scale |


|
Download:
|
| Fig. 2. Photographs of the reaction flask after the reaction (a) and the purified product 3a (b) | |
As (E)-4-phenylbut-3-en-2-one (1a) was synthesized through the Ca(OH)2-catalyzed condensation of benzaldehyde (2a) with acetone, we then tried to combined the two reaction steps in one pot to prepare dialkylideneacetone 3a. In the process, 2a first reacted with acetone to produce 1a. The excess acetone and solvent were then removed by distillation and another solution of benzaldehyde 2a in fresh EtOH/H2O was added. After heating at 80 ℃ for 48 h, the product 3a precipitated and could be isolated by filtration and purified by washing with water (Table 3, entry 1). Other dialkylideneacetones 3b–i were smoothly prepared in similar way (Table 3, entries 2–9).
|
|
Table 3 One-pot synthesis of dialkylideneacetone 3 from 2a |

Moreover, the order of introducing functional groups was reversible. As shown in Table 4, substituted aldehydes 2 first reacted with acetone to generate the intermediate 1 in situ, which could then react with another benzaldehyde (2a) to produce the same products 3 in moderate to excellent yields (Table 4, entries 1– 8). Notably, the yields of 3b–c could be enhanced by using this protocol (Table 4, entries 1 and 2 vs. Table 3, entries 2 and 3).
|
|
Table 4 One-pot synthesis of dialkylideneacetone 3 from substituted aldehydes 2 |

{kind=link}
{kind=link}
{kind=link}
In conclusion, we developed a practical method for the synthesis of dialkylideneacetones through the Ca(OH)2-catalyzed Claisen-Schmidt condensations in dilute ethanol. The cheap, lowloading and mild catalyst, green solvent and the easily-operated procedures are the advantages of the protocol over many known methods. The reaction was scalable to at least 100 mmol to produce the desired dialkylideneacetones in grams, showing very good practicability for both laboratory synthesis as well as the industrial level production.
AcknowledgmentsWe thank the National Natural Science Foundation of China (Nos. 21202141, 21672163), Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), Top-notch Academic Programs Project of Jiangsu Higher Education Institutions (TAPP), the high level talent support project of Yangzhou University (top-notch talent, L. Yu), Yangzhou Natural Science Foundation (No. YZ2014040) and the Natural Science Foundation of Guangling College (No. ZKZD17005) for financial support.
| [1] |
J. Deli, T. Lorand, D. Szabo, A. Foldesi, Pharmazie 39 (1984) 539-540. |
| [2] |
N.J. Leonard, L.A. Miller, J.W. Berry, J. Am. Chem. Soc. 79 (1957) 1482-1485. DOI:10.1021/ja01563a056 |
| [3] |
M.A. Ciufolini, N.E. Byrne, J. Am. Chem. Soc. 113 (1991) 8016-8024. DOI:10.1021/ja00021a031 |
| [4] |
T.P. Robinson, T. Ehlers, R.B. Hubbard, et al., Bioorg. Med. Chem. Lett. 13 (2003) 115-117. DOI:10.1016/S0960-894X(02)00832-6 |
| [5] |
T.P. Robinson, R.B. Hubbard, T.J. Ehlers, et al., Bioorg. Med. Chem. 13 (2005) 4007-4013. |
| [6] |
A.T. Dinkova-Kostova, C. Abeygunawardana, P. Talalay, J. Med. Chem. 41 (1998) 5287-5296. DOI:10.1021/jm980424s |
| [7] |
D. Cheng, S. Valente, S. Castellano, et al., J. Med. Chem. 54 (2011) 4928-4932. DOI:10.1021/jm200453n |
| [8] |
K.D. Ashtekar, X. Ding, E. Toma, et al., Org. Lett. 18 (2016) 3976-3979. DOI:10.1021/acs.orglett.6b01742 |
| [9] |
A.J. Reay, L.K. Neumann, I.J.S. Fairlamb, Synlett 27 (2016) 1211-1216. DOI:10.1055/s-00000083 |
| [10] |
N. Castellanos-Blanco, M. Flores-Alamo, J.J. García, Organometallics 31 (2012) 680-686. DOI:10.1021/om2010222 |
| [11] |
L.L. Franco, M.V. de Almeida, L.F.R. e Silva, et al., Chem. Biol. Drug Des. 79 (2012) 790-797. DOI:10.1111/jpp.2012.79.issue-5 |
| [12] |
K.K. Gangadhara, Polymer 36 (1995) 1903-1910. DOI:10.1016/0032-3861(95)90938-X |
| [13] |
G. Aullón, P. Romea, F. Urpí, Synthesis 49 (2017) 484-503. |
| [14] |
W. Gati, H. Yamamoto, Acc. Chem. Res. 49 (2016) 1757-1768. DOI:10.1021/acs.accounts.6b00243 |
| [15] |
A.F.M.M. Rahman, R. Ali, Y. Jahng, A.A. Kadi, Molecules 17 (2012) 571-583. DOI:10.3390/molecules17010571 |
| [16] |
Z.X. Shan, X.X. Luo, L. Hu, X.Y. Hu, Sci. China:Chem. 53 (2010) 1095-1011. DOI:10.1007/s11426-010-0137-5 |
| [17] |
Y.F. Sun, Z.Y. Wang, X. Zhao, et al., Dyes Pigm. 86 (2005) 97-105. |
| [18] |
C.L. Raston, J.L. Scott, Green Chem. 2 (2000) 49-52. DOI:10.1039/a907688c |
| [19] |
E.L. Gall, F. Texier-Boullet, J. Hamelin, Synth. Commun. 29 (1999) 3651-3657. DOI:10.1080/00397919908086000 |
| [20] |
J. Li, W. Su, N. Li, Synth Commun. 35 (2005) 3037-3043. DOI:10.1080/00397910500278735 |
| [21] |
X. Zhang, X. Fan, H. Niu, J. Wang, Green Chem. 5 (2003) 267-269. DOI:10.1039/b212155g |
| [22] |
N. Iranpoor, B. Zeynizadeh, A. Aghapour, J. Chem. Res. (Synopses) 9 (1999) 554-555. |
| [23] |
X. Zheng, Y. Zhang, Synth. Commun. 33 (2003) 161-165. DOI:10.1081/SCC-120015572 |
| [24] |
L. Wang, J. Sheng, H. Tian, et al., Synthesis 18 (2004) 3060-3064. |
| [25] |
N. Iranpoor, F. Kazemi, Tetrahedron 54 (1998) 9475-9480. DOI:10.1016/S0040-4020(98)00575-4 |
| [26] |
Y. Zhu, Y. Pan, Chem. Lett. 33 (2004) 668-669. DOI:10.1246/cl.2004.668 |
| [27] |
J.S. Yadav, B.V.S. Reddy, A. Nagaraju, J.A.R.P. Sarma, Synth. Commun. 32 (2002) 893-896. DOI:10.1081/SCC-120002700 |
| [28] |
A. Hasaninejad, A. Zare, L. Balooty, M. Mehregan, M. Shekouhy, Synth. Commun. 40 (2010) 3488-3495. DOI:10.1080/00397910903457282 |
| [29] |
T. Wang, X. Jing, C. Chen, L. Yu, J. Org. Chem. 82 (2017) 9342-9349. DOI:10.1021/acs.joc.7b01245 |
| [30] |
D. Zhang, Z. Wei, L. Yu, Sci. Bull. 62 (2017) 1325-1330. DOI:10.1016/j.scib.2017.09.016 |
| [31] |
X. Jing, D. Yuan, L. Yu, Adv. Synth. Catal. 359 (2017) 1194-1201. DOI:10.1002/adsc.v359.7 |
| [32] |
F. Wang, L. Xu, J. Huang, et al., Mol. Catal. 432 (2017) 99-103. DOI:10.1016/j.mcat.2017.02.010 |
| [33] |
Y. Wang, L. Yu, B. Zhu, L. Yu, J. Mater. Chem. A 4 (2016) 10828-10833. DOI:10.1039/C6TA02566H |