 2019, Vol. 30
2019, Vol. 30 

 ,
Chunquan Shenga,*,
Jianzhong Yaoa,*
,
Chunquan Shenga,*,
Jianzhong Yaoa,*
b Department of Pharmacy, 455 th Hospital of Chinese People's Liberation Army, Shanghai 200052, China;
c Department of Radiology, Shanghai Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, Shanghai 200438 China
Photodynamic therapy (PDT) now is an attractive approach to innovative cancer therapy involving combined use of visible light and a photosensitizer (PS) [1]. PDT relies on the interaction between light and PS in tumor tissues to generate superoxide anions and radicals (type Ⅰ reaction) or highly cytotoxic singlet oxygen (type Ⅱ reaction) with the ultimate formation of reactive oxygen species (ROS) to inactivate the tumor cells [2].
Porfimer sodium, the first generation of porphyrin-type PS, has achieved enormous clinical efficacy for the treatment of bladder cancer in the world. It also has suffered from some serious drawbacks such as complex component, poor tissue penetration due to its limited maximum absorption wavelength of 630 nm, inefficient absorption (ε = 1170 L mol-1 cm-1) at 630 nm, and prolonged cutaneous phototoxicity up to 4-6 weeks after treatment caused by its slow elimination in skin tissues [3].
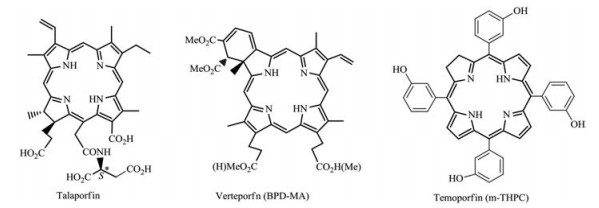
A great number of so-called the second generation of PSs especially related to chlorins such as chlorophyll-a derivatives etc., are becoming more and more concerned owing to rapid clearance from tissues and intense absorption in near-infrared region (> 650 nm, also called "phototherapeutic window"), which are relatively harmless and penetrate deeply in biological tissues [4-6]. Among them, talaporfin [7], verteporfin (BPD-MA) [8] and temoporfin (m-THPC) [9] were clinically approved for PDT applications (Fig. 1).

|
Download:
|
| Fig. 1. Three clinical available chlorin-type photosensitizers | |
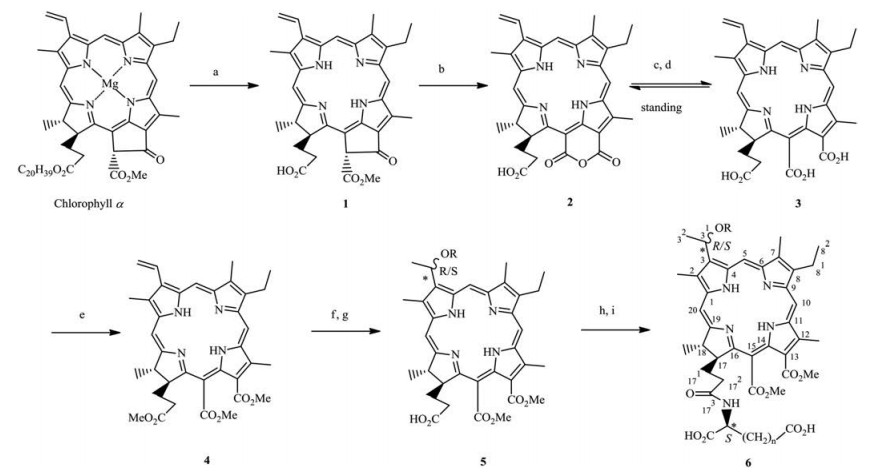
Chlorin p6 (3), the one of chlorophyll-a derivatives as chlorintype PS, has poor stability to hamper its clinical development because it is easily converted into stable purpurin-18 (2) with poor water solubility by its automatically intramolecular dehydration in the neutral condition. Chlorin p6 trimethylester (4), which is formed by methylation of 3, has good stability but poor water solubility. Because introducing amino acid was reported to be an effective strategy to improve the water-solubility and the biological activity of chlorin- and porphyrin-based derivatives [10-12], we previously synthesized some chlorin p6- and pyropheophorbide-a-based water-soluble amino acid derivatives and obtained a candidate PS with a better efficacy than verteporfin [13, 14]. Considering that alkoxyl ether derivatives of chlorin at 31-position exhibited stronger photosensitive activity thanparent compound [15], a series of novel water-soluble amino acid conjugates 6a-h of chlorine p6 ethers (5a-d) were further designed, synthesized and preliminarily investigated their photodynamic antitumor activity against melanoma B16-F10 and mammary carcinoma cells (Scheme 1).

|
Download:
|
| Scheme 1. Synthetic route for the titled compounds 6a-h. Reagents and conditions: (a) cond. aqueous HCl-Et2O, 0–5 ℃, 30 min; (b) KOH, i-PrOH-Et2O, 12 h, 34.4%; (c) THFCH3OH- aqueous NaOH (0.5 mol/L) (1:4:5, v/v/v), r.t., 1 h; (d) 0.5 mol/L aqueous HCl to adjust pH value to 5-6, dilution with H2O, extraction with Et2O, dried by Na2SO4 for 1 h; (e) CH2N2, 83.6% (from c to e); (f) 33% HBr-HOAc, r.t., 36 h; (g) alcohol, CH2Cl2, K2CO3, r.t., 2.5 h. Alcohol donors: R = CH3 (5a), n-C3H7 (5b), n-C4H9 (5c), n-C5H11 (5d); (h) L-(S)- (+)-Asp(OBut)2·HCl (n = 1) or L-(S)-(+)-Glu(OBut)2·HCl (n = 2), EDC·HCl, HOBt, DIPEA, CH2Cl2, r.t., 12 h; (i) CH2Cl2-TFA (3:1), r.t., 6 h. Alcohol donors and amino acid residues: n = 1 and R = CH3 (6a), n-C3H7 (6b), n-C4H9 (6c), n-C5H11 (6d); n = 2 and R = CH3 (6e), n-C3H7 (6f), n-C4H9 (6g), n-C5H11 (6h). | |
As shown in Scheme 1, all intermediates pheophorbide-a (1), purpurin-18 (2) and chlorin p6 trimethylester (4) were obtained via acid and base degradation of chlorophyll-a followed by carboxyl methylation according to our previous methodology developed in our laboratory using crude chlorophyll extracts in Chinese traditional herb named silkworm excrements [16, 17]. Briefly, intermediate 1 was got via cond. aqueous HCl degradation of chlorophyll-a in Et2O. Treatment of 1 in Et2O with KOH-i-PrOH under an atmosphere of O2 gave 2 in 34.4% yield. 13, 15-Anhydride ring of 2 was hydrolyzed in the presence of tetrahydrofuran (THF) and CH3OH using NaOH as the base to form unstable chlorin 3, which was rapidly methylated in Et2O with CH2N2 to give 4 in 83.6% yield from 2.
In this paper, the details of the synthesis of key intermediate 5a-d and target compounds 6a-h from initial intermediate 4 were also given in Supporting information. Briefly, addition of 4 with 33% HBr in HOAc followed by substitution with excessive alcohol donors (ROH) in the presence of K2CO3 produced chlorin p6 ether derivatives 3-devinyl-3-(1-(R/S)-alkoxy)ethyl-chlorin p6-13, 15-dimethylester (5a-d) in modest yields ranged from 26.7% to 32.7%. Obviously, intermediate 5 consisted of two epimerides of R- and S-configuration at 31-alkoxyl. High performance liquid chromatography (HPLC) analysis with chiral column showed that the general peak area ratio of the two epimerides was individually 22.5% vs. 77.5% for 5a, 35.9% vs. 64.1% for 5b, 31.3% vs. 68.7% for 5c, 33.3% vs. 65.7% for 5d (Figs. S1–S4 in Supporting information). Then, key intermediate 5a-d was each coupled with L-(S)- (+)-aspartic acid hydrochlorate or L-(S)-(+)-glutamic acid hydrochlorate whose carboxyl protected by tert-butyl in the presence of 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochlorate (EDC·HCl), 1-hydroxybenzotriazole (HOBt) and N, N-diisopropylethylamine (DIPEA) followed by removal of tert-butyl with trifluoroacetic acid (TFA) to generate the target compounds N-(3-devinyl-3-(1-(R/S)-alkoxy)ethyl-chlorin p6-13, 15-dimethylester-173-acyl)-L-(S)-(+)-aspartic acid (6a-e) and N-(3-devinyl-3- (1-(R/S)-alkoxy)ethyl-chlorin p6-13, 15-dimethyl ester-173-acyl)-L- (S)-(+)-glutamic acid (6f-h) in receivable yields ranged from 50.0% to 77.1%. Similarly, target compound 6 also was composed of two anisometric epimerides of R- and S-configuration at 31-alkoxyl, which possessed characteristic of dextral rotation (Supporting information), and its general peak area ratio of two epimerides analyzed by HPLC with chiral column was 35.2% vs. 64.8% for 6c, 35.3% vs. 64.7% for 6d, 41.8% vs. 58.2% for 6g, 48.2% vs. 51.8% for 6h, respectively (Figs. S5–S8 in Supporting information). It is limited to our technical conditions that a pair of epimers in both 5 and 6 was even failure to complete effective separation and preparation. In general, the change of minor group configuration has generally little effect on the PDT antitumor activity as PS belongs to structural nonspecific drugs without specific drug target such as enzymes, receptors and proteins etc. The structures of key intermediate 5a-d and target compounds 6a-h were identified by 1H NMR, 13C NMR, ESIMS and elemental analysis data (Supporting information). In addition, the UV-visible spectral data showed that they possessed more efficient absorption (ε = 1.67 × 105-2.42 ×105 L mol-1 cm-1) at longer maximum absorption wavelength of 660–662 nm than porfimer sodium (ε = 1170 L mol-1 cm-1) at 630 nm (Table 1), suggesting their greater tissue penetration [4-6].
|
|
Table 1 UV-vis data of the synthetic titled compounds |
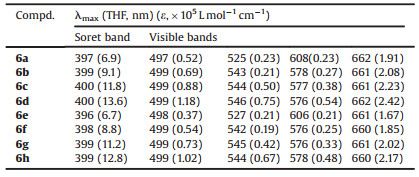
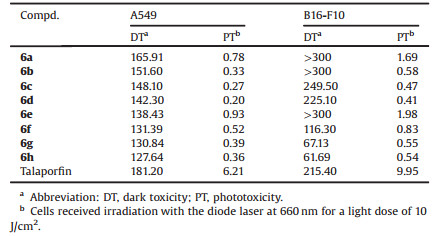
In order to evaluate the effect of the introduced different amino acid moiety at 173-carboxyl and alkoxyl at 31-alkoxyl on the growth inhibitory activity of the target compounds against human cancer cell lines and to clarify the SAR, the dark toxicity and phototoxicity of all the target compounds 6a-h were measured by the CCK-8 assay against human mammary carcinoma and murine melanoma B16-F10 cells using talaporfin as positive control (Supporting information). To eliminate the experimental error caused by solvent, all tested compounds and talaporfin were both made into water-soluble sodium salt. As shown in Table 2, all the compounds exhibited better phototoxicity and considerable dark toxicity against two tested tumor cell lines compared to talaporfin. SAR analysis showed that their PDT antitumor activity enhanced with the increase of carbon chain length of alkoxyl ether bonds at 31-position, and L-aspartic acid was superior to L-glutamic acid. Among them, compound 6d exhibited the best in vitro PDT antitumor efficacy and its IC50 values against A549 and B16-F10 cells were individually 0.20 μmol/L and 0.41 μmol/L, which represented 31- and 24-fold increase of antitumor potency compared to talaporfin, respectively.
|
|
Table 2 Cytotoxicity (IC50, μmol/L) for the titled compounds against tumor cells |
{kind=link}
{kind=link}
In summary, 8 new water-soluble amino acid conjugates 6a-h of 31-alkoxyl ethers (5a-d) for chlorin p6 trimethylester (4) were synthesized and evaluated for their preliminary in vitro photodynamic antitumor activity against A549 and B16-F10 cells. All target compounds exhibited much stronger phototoxicity against tested tumor cell lines than talaporfin. In particular, compounds 6d was the most effective, which individually showed 31- and 24-fold antitumor potency on A549 and B16-F10 cells compared to talaporfin. As a result, compound 6d represents a promising PS for PDT applications owing to its strong absorption at long wavelength, high phototoxicity, low dark cytotoxicity and good water-solubility. Moreover, its more extensive and deeper physical and biological study including singlet oxygen quantum yield, in vivo PDT antitumor efficacy, subcellular localization and tumor cell apoptosis detection etc. are ongoing.
AcknowledgmentsThis work was supported by grants from the National Natural Science Foundation of China (Nos. 81172950 and 81671739), and the Project of Science and Technology Commission of Shanghai (No. 11431920401) and the College Students' Innovation Ability Training Project of Second Military Medical University (No. MS2017040).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2018.04.029.
| [1] |
P. Agostinis, K. Berg, K.A. Cengel, et al., Cancer J. Clin. 61 (2011) 250-281. DOI:10.3322/caac.v61:4 |
| [2] |
R.W. Redmond, I.E. Kochevar, Photochem. Photobiol. 82 (2006) 1178-1186. DOI:10.1562/2006-04-14-IR-874 |
| [3] |
N. Drogat, C. Gady, R. Granet, V. Sol, Dyes Pigment. 98 (2013) 609-614. DOI:10.1016/j.dyepig.2013.03.018 |
| [4] |
K.T. Oliveira, P.B. Momo, F.F. Assis, et al., Curr. Org. Synth. 11 (2014) 42-58. DOI:10.2174/15701794113106660085 |
| [5] |
J.Z. Yao, W.N. Zhang, C.Q. Sheng, et al., Bioorg. Med. Chem. Lett. 18 (2008) 293-297. DOI:10.1016/j.bmcl.2007.10.086 |
| [6] |
J.L. Zhang, L. Deng, J.Z. Yao, et al., Bioorg. Med. Chem. 19 (2011) 5520-5528. |
| [7] |
D. Kessel, Photochem. Photobiol. 49 (2008) 447-452. |
| [8] |
R.R. Allison, G.H. Downie, R. Cuenca, et al., Photodiagn. Photodyn. 1 (2004) 27-42. DOI:10.1016/S1572-1000(04)00007-9 |
| [9] |
H. Abrahamse, M.R. Hamblin, J. Biochem, 473 (2016) 347-364.
|
| [10] |
T. Hitoshi, I. Yasuaki, T. Takuya, et al., Bioorg. Med. Chem. 22 (2014) 1421-1428. |
| [11] |
W.G. Roberts, F.Y. Shiau, J.S. Nelson, et al., J. Natl. Cancer Inst. 80 (1988) 330-336. DOI:10.1093/jnci/80.5.330 |
| [12] |
M. Kwitniewski, A. Juzeniene, L.W. Ma, et al., J. Photochem. Photobiol. B 94 (2009) 214-222. DOI:10.1016/j.jphotobiol.2008.11.005 |
| [13] |
Z. Meng, B. Yu, G.Y. Han, et al., J. Med. Chem. 59 (2016) 4999-5010. DOI:10.1021/acs.jmedchem.6b00352 |
| [14] |
Z. Meng, B. Shan, L. Zhang, et al., Chin. Chem. Lett. 27 (2016) 623-626. DOI:10.1016/j.cclet.2016.03.019 |
| [15] |
J.Z. Yao, W.H. Chen, X. He, et al., Acta Pharm. Sin. 35 (2000) 63-66. |
| [16] |
J.Z. Yao, D.Y. Xu, W.H. Chen, et al., Chin. J. Pharm. 30 (1999) 403-406. |
| [17] |
J.Z. Yao, J.F. Liu, W.N. Zhang, et al., Chin. J. Org. Chem. 21 (2001) 458-462. |