 2019, Vol. 30
2019, Vol. 30 


At present, the use of fossil energy has caused great environmental pollution, thus it is imperative to explore economical renewable substitute for fossil energy which occupies the main position in the overall consumption of energy in the world [1, 2]. Hydrogen (H2) is considered to be an ideal candidate to replace fossil fuels, as it has the advantages of highest gravimetric energy density and zero net greenhouse gas emissions [3]. Among the multitudinous researches to produce clean and sustainable hydrogen energy, electrochemical water splitting is an ideal approach owing to its environment-friendly mode of production [4, 5]. However, due to the sluggish kinetics and the need of high overpotential to drive the four-electron reaction, the anodic oxygen evolution reaction (OER) is an obstacle for the commercialized application of electrochemical water splitting [6]. While IrO2 and RuO2 manifest high catalytic activity for OER, the high cost stop them from extensive applications [7]. Therefore, a lot of researches have focused on the development of efficient and abundant OER electrocatalysts.
In recent decades, many potential alternatives including metal oxides [8, 9], metal hydroxides [10], metal phosphates [11, 12], metal sulfides [13, 14], metal carbides [15], metal nitrides [16], metal selenides [17, 18], perovskites [19] and carbon-based catalysts [20, 21], etc., have been identified as efficient electrocatalysts for OER. Among all these alternatives, transition metal sulfides have been regarded as promising electrocatalytic materials since these materials have a good balance between catalytic activity, durability, storage and cost [22, 23]. However, there is still a lot of room to reduce the overpotential and improve reaction kinetics for OER. A large number of modified methods have been proposed, such as constructing nanostructure to obtain large active surface area [13, 24], combining catalysts with conductive supports to accelerate the electron transfer rate [14, 25], doping to tuning the structure of electronic states [26], etc. Coupling transition metal sulfides with carbon support is a widely adopted method owing to its potential in increasing the conductivityand dispersing catalysts homogeneously [27, 28], which can improve the OER electrocatalytic properties. Yang and co-workers found that combining high efficiency OER catalyst with conductive graphite substrate exposed more catalytically active sites, thereby improving the charge transport as well as catalytic activity [29]. Shanmugam and co-workers reported that the excellent electrocatalytic performance of cobalt sulfide and graphene composite catalyst could be attribute to uniform dispersion suitable anchoring of CoS2 particles on the graphene [27].
Metal-organic frameworks (MOFs), yielded from the selfassembly of metal cations or clusters and organic ligands through coordination bonds [30, 31], have been regarded as alternative precursors to synthesize electrocatalysts due to their ordered crystalline structures, high porosity with controlled pore sizes, modifiable functionality and large surface area [32, 33]. Porous structure of catalysts is helpful for OER performance, as it can provide rich edge active sites and high surface area for promoting the reaction rate [34], as well as abundant pores for accelerating the circulation of electrolyte and the release of gas. MOFs are ideal precursors to fabricate porous materials, the pores can be introduced by simple heat treatment due to the volatilization of small molecular gases. In particular, with rich metal and carbon atoms, MOFs are frequently used to synthesize carbon supported metal-based nanocomposites [35]. As a highly porous Co-based zeolitic imidazolate framework, ZIF-67 has been advisedly chosen to design carbon supported Co-based hybrid materials [36, 37]. For examples, Tang and co-workers fabricated a composite of defect-rich CoP and nitrogen-doped carbon through low-temperature phosphorization of ZIF-67, which performed efficient hydrogen evolution reaction property [38]. Xia and co-workers reported the preparation of highly homogeneous dispersed cobalt sulfide nanoparticles incorporated in N, S co-doped porous carbon by simple sulfurization and carbonization of ZIF-67, and this composite showed excellent electrocatalytic activity towards OER [35].
In this work, we reported a two-step conversion strategy to prepare a novel composite catalyst of Co1-xS embedded in porous carbon framework (Co1-xS@C). After being synthesized by a solution-precipitation method, the ZIF-67 precursor was transformed into porous dodecahedron Co1-xS@C catalyst through oxidation and sulfurization, successively. The Co1-xS@C composite catalyst obtained by this simple and economical fabrication process, exhibited excellent catalytic activity and stability for OER. It only needed an overpotential of 260 mV to achieve 10 mA/cm2 for OER in 1.0 mol/L KOH with a Tafel slope of 85 mV/dec. Moreover, a long-term stability over 80 h was observed under galvanostatic measurement. This work provided a cost-effective and scalable strategy for the design of earth-abundant hybrid electrocatalysts derived from MOFs.
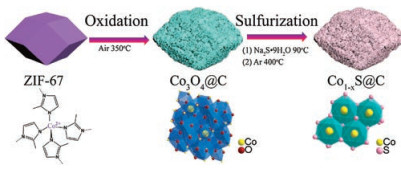
The conversion process of the porous dodecahedra Co1-xS@C from ZIF-67 is illustrated in Fig. 1. Prior to conversion, the ZIF-67 precursor with rhombic dodecahedral morphology was synthesized via the coordination of Co2+ cations and 2-methylimidazolate by a reported method [39]. The synthesis of porous dodecahedron Co1-xS@C composite catalyst involved two typical steps. In the first step, the purple ZIF-67 powders were directly calcined at 350 ℃ in air. The obtained product was denoted as Co3O4@C. In the second step, Co3O4@C was transformed to amorphous cobalt sulfide (denoted as amorphous CoSx@C) through ion-exchange [40], then Co1-xS@C was obtained by calcining the amorphous CoSx@C at 400 ℃ in Ar.

|
Download:
|
| Fig. 1. Schematic illustration of the synthesis procedure of Co1_xS@C. | |
{kind=link}
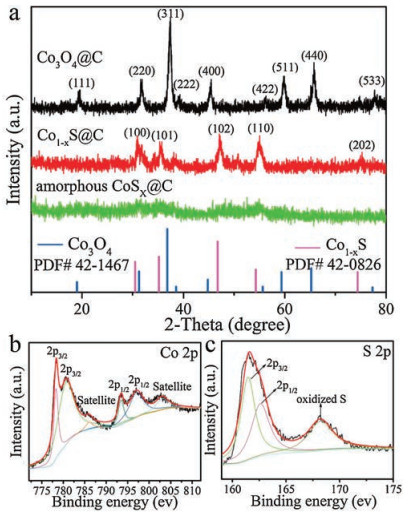
As exhibited in Fig. 2a, the crystal structures of the prepared samples were verified by X-ray diffraction (XRD). The diffraction peaks of Co3O4@C were well matched with corresponding standard diffraction patterns of cubic Co3O4 (JCPDS No. 42-1467) [41], which suggested the formation of cubic structured Co3O4 after oxidation process. No obvious peak emerged in the XRD patterns of amorphous CoSx@C, indicating that an amorphous product was obtained by ion-exchange. After crystallization at 400 ℃, the diffraction peaks of Co1-xS at 30.5°, 35.1°, 46.8°, 54.3° and 74.4° can be indexed to (100), (101), (102), (110) and (202) crystal faces of hexagonal Co1-xS (JCPDS No. 42-0826) [42].

|
Download:
|
| Fig. 2. (a) XRD patterns of Co3O4@C, amorphous CoSx@C and Co1-xS@C. XPS spectra of the porous dodecahedra Co1-xS@C for (a) Co 2p, and (b) S 2p. | |
{kind=link}
X-ray photoelectron spectroscopy (XPS) was used to further analyze the chemical composition and valence of Co1-xS@C. As shown in Fig. 2b, the Co 2p spectrum was deconvoluted into Co 2p1/2 and Co 2p3/2 spin-oribit coupling, accompanying with weak satellite peaks. The peaks located at 778.3 eV and 793.5 eV, 780.6 eV and 797.0 eV indicate the co-existence of Co2+ and Co3+ cations in Co1-xS@C [43, 44]. For S 2p, the peaks centered at 161.4 eV and 162.6eV are assigned to S 2p1/2 and 2p3/2 of S2— in Co1-xS as shown in Fig. 2c [45], and another peak at 168.2eV corresponding to the oxidized S may be caused by oxidation in the air. The C 1 s spectrum of Co1-xS@C was displayed in Fig. S1 (Supporting information). Furthermore, the atomic ratio of Co and S obtained by XPS is 0.664 and the corresponding x value of Co1-xS@C is 0.336, which conforms to the compositional range 0 ≤ x ≤ 0.5 of reported literature [46].
Scanning electron microscopy (SEM) and transition electron microscopy (TEM) were performed to further illustrate the microstructure and morphology of the product at each stage. Rhombic dodecahedral structures with flat smooth surface of ZIF-67 can be seen in Fig. 3a, and the average size is about 150 ~200nm. As shown in Fig. 3b, after the two-step conversion, the obtained Co1-xS@C exhibited hollow dodecahedral structure and honeycomb-like porous morphology, almost remained the microstructure features when compared to ZIF-67 precursor. The SEM images of Co3O4@C and amorphous CoSx@C (Fig. S2 in Supporting information) also proved that the morphology and microstructure remain unchanged. TEM image in Fig. 3c reveals an approximate hexagonal projection of porous dodecahedron Co1-xS@C, which is in conformity with the SEM image. TEM images of ZIF-67, Co3O4@C and Co1-xS@C are displayed together for comparison (Fig. S3 in Supporting information), showing that the rhombic dodecahedron does not change too much during the oxidation and sulfurization process. Fig. 3d demonstrates the high resolution TEM (HRTEM) investigation of Co1-xS@C, the crystal lattice fringe with inter-planar distance of 0.194 nm matches well with the XRD dominant peak (102) of Co1-xS. As shown in Fig. 3e, energy dispersive X-ray spectroscopy (EDS) was performed to explore the spatial distribution of different elements. It is clearly that Co and S elements distribute evenly in the whole porous dodecahedron. Even though the ultrathin carbon support membrane exerts an influence on the detection result of carbon element, a high density of C element distribution can be easily seen emerging on the hexagonal region, proving the existence of C element on the framework.

|
Download:
|
| Fig. 3. (a) SEM image of ZIF-67, (b) SEM image of Co1-xS@C, (c) TEM image of Co1-xS@C, (d) HRTEM image of Co1-xS@C and the inset shows the corresponding FFT pattern. (e) EDS elemental mapping images of Co1-xS@C. | |
{kind=link}
The N2 adsorption/desorption measurements were carried out to explore the specific surface area and pore structures of Co1-xS@C. The similar adsorption/desorption curves and pore size distribution plots proved the presence of rich mesopores in the porous dodecahedra Co1-xS@C (Fig. S4 in Supporting information). The similar N2 adsorption/desorption curves and pore size distribution of Co1-xS@C and Co3O4@C indicate that the pore structures remain nearly constant through the mild sulfurization process. Generally speaking, the proposed two-step conversion method for synthesizing porous sulfide from MOFs is contributed to the maintenance of polyhedron morphology and porous structure, which could expose more active sites and promote mass transfer during electrochemical catalytic process.
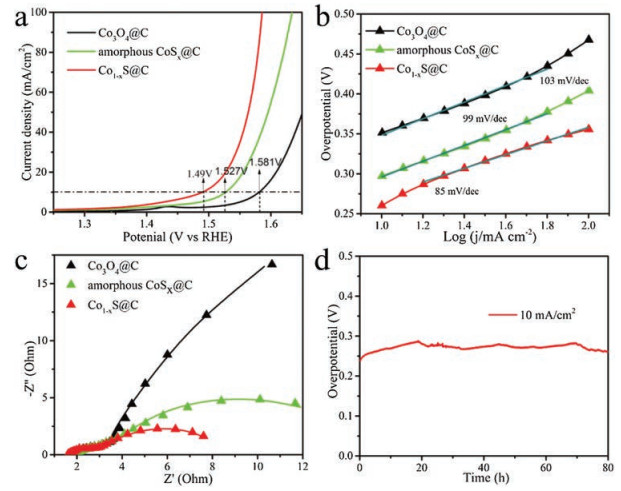
To investigate electrochemical catalytic activity of these Cobased materials, the OER performances were studied by using a standard three-electrode setup. The iR-corrected polarization curves were examined by linear sweep voltammetry (LSV) as shown in Fig. 4a, with low overpotential of 260 mV to achieve 10mA/cm2, Co1-xS@C demonstrated excellent OER activity when compared to Co3O4@C and amorphous CoSx@C (351 mV and 297 mV, respectively). In order to explore the influence of crystallization temperature on catalytic performance, the amorphous CoSx@C obtained by sulfurization were calcined under different temperatures (350, 400 and 450 ℃) and tested for comparison (Fig. S5 in Supporting information). The Co1-xS@C calcined at 400 ℃ showed better catalytic activity towards OER than the other two catalysts calcined at 350 ℃ and 450 ℃. The XRD and SEM images (Figs. S6 and S7 in Supporting information) suggested that 350 ℃ is not enough for complete crystallization, but 450 ℃ is so high that it converts to other sulphides of cobalt. Then a conclusion could be drawn that the sample crystallized at 400 ℃ can not only maintain the porous dodecahedra structure and morphology, but also achieve high crystallinity, thus it showed the best electrochemical catalytic performance. Tafel slopes were displayed in Fig. 4b to discuss the kinetic mechanism for OER, Co1-xS@C exhibited a Tafel slope value ofb = 85 mV/dec, which was lower than Co3O4@C and amorphous CoSx@C (103 and 99 mV/dec, respectively), suggesting the faster OER kinetics over Co1-xS@C catalyst. To prove the excellent catalytic activity of Co1-xS@C, the electrochemical impedance spectroscopy (EIS) was used to explore the electrode kinetics under OER process as shown in Fig. 4c. In accordance with the trend of Tafel slopes, the Nyquist plots revealed a dramatic decrease of Co1-xS@C compared to Co3O4@C and amorphous CoSx@C, also proving enhanced electron transfer rate and faster catalytic kinetics of Co1-xS@C towards OER. It can be concluded that Co1-xS@C exhibits the excellent OER catalytic activity in low overpotential at 10 mA/cm2, low Tafel slope and fast electron transfer rate. Moreover, Co1-xS@C indicated competitive OER performance to other materials derived from MOFs or transition-metal sulfides (Table S1 in Supporting information). These enhancements are owing to its porous dodecahedra structure with ample available electrochemical active sites and the better electron conductivity of metal sulfides than metal oxides [40].

|
Download:
|
| Fig. 4. (a) iR-compensated LSV polarization curves and (b) the corresponding Tafel plots of Co3O4@C, amorphous CoSx@C and Co1-xS@C. (c) EIS Nyquist plots and fitted solid traces of Co3O4@C, amorphous CoSx@C and Co1-xS@C at an OER overpotential of 370 mV. (d) Chronopotentiometric measurement of Co1-xS@C at -10 mA/cm2. All of these electrochemical tests were performed in 1.0 mol/L KOH (pH 13.6). | |
{kind=link}
The electrochemical double layer capacitance (Cdl) of materials is proportional to its effective surface area [47].The Cdl of Co3O4@C, amorphous CoSx@C and Co1-xS@C is 58.4, 61.2 and 74.2 mF/cm2, respectively (Fig. S8 in Supporting information). This remarkable difference of Cdl implies that the excellent OER performance of Co1-xS@C may attribute to the larger effective surface area enabled by the high porosity and good conductivity. The stability was investigated by performing a continuous galvanostatic measurement at 10mA/cm2 As observed in Fig. 4d, the overpotential exhibits only a slight change from 260 mV even after a long test of 80 h, suggesting the excellent durability of Co1-xS@C towards oxygen evolution. Furthermore, we also studied the Faradaic efficiency of O2 produced by the porous Co1-xS@C anode (Fig. S9 in Supporting information). The value of Faradaic efficiency closes to 100%, suggesting that there is less side reaction and Co1-xS@C is an excellent catalyst for OER.
In conclusion, we have developed a facile strategy to construct Co1-xS@C composite OER electrocatalyst by a two-step conversion of ZIF-67. The porous dodecahedra Co1-xS@C exhibited efficient OER activity with low overpotential of 260 mV (10 mA/cm2), small Tafel slope of ~85mV/dec and long-term durability over 80h in alkaline solution. The excellent performance of this composite catalyst may owe to the prominent conductivity of Co1-xS, the porous structure for promoting mass transfer, and uniform dispersion of Co1-xS particles in carbon framework for exposing adequate catalytical active sites. In general, with the great advantages of various metal atoms and rich carbon atoms, and porous and tunable structure, MOFs will undoubtedly become promising precursors or platforms for the synthesis of carbon based composite catalysts. And the two-step conversion method is helpful to maintain the morphology and structure of MOFs during the conversion process. We believe that such a synthetic strategy shows the potential to prepare other composite materials for various applications.
AcknowledgmentsThis research was supported by China Major Science and Technology Program for Water Pollution Control and Treatment (No. 2017ZX07101003), Zhejiang Provincial Natural Science Foundation of China (No. LR17B060003). The work was also financially supported by the National Science Foundation of China (Nos. 21436007, 21522606, 21476201, 21676246, U1462201, and 21776248).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2018.03.020.
| [1] |
Y. Na, B. Hu, Q.L. Yang, et al., Chin. Chem. Lett. 26 (2015) 141-144. DOI:10.1016/j.cclet.2014.09.011 |
| [2] |
X. Zou, Y. Zhang, Chem. Soc. Rev. 44 (2015) 5148-5180. DOI:10.1039/C4CS00448E |
| [3] |
P.E. Dodds, I. Staffell, A.D. Hawkes, et al., Int. J. Hydrogen Energy 40 (2015) 2065-2083. DOI:10.1016/j.ijhydene.2014.11.059 |
| [4] |
L.A. Stern, L. Feng, F. Song, et al., Energy Environ. Sci. 8 (2015) 2347-2351. DOI:10.1039/C5EE01155H |
| [5] |
D.J. Li, U.N. Maiti, J. Lim, et al., Nano Lett. 14 (2014) 1228-1233. DOI:10.1021/nl404108a |
| [6] |
C. Tang, H.S. Wang, H.F. Wang, et al., Adv. Mater. 27 (2015) 4516-4522. DOI:10.1002/adma.v27.30 |
| [7] |
Y. Dou, T. Liao, Z. Ma, et al., Nano Energy 30 (2016) 267-275. DOI:10.1016/j.nanoen.2016.10.020 |
| [8] |
L. Xu, Q. Jiang, Z. Xiao, et al., Angew. Chem. 128 (2016) 5363-5367. DOI:10.1002/ange.201600687 |
| [9] |
Z. Xiao, Y. Wang, Y.C. Huang, et al., Energy Environ. Sci. 10 (2017) 2563-2569. DOI:10.1039/C7EE01917C |
| [10] |
J. Jiang, A. Zhang, L. Li, L. Ai, J. Power Sources 278 (2015) 445-451. DOI:10.1016/j.jpowsour.2014.12.085 |
| [11] |
X.Y. Yu, Y. Feng, B. Guan, X.W. Lou, U. Paik, Energy Environ. Sci. 9 (2016) 1246-1250. DOI:10.1039/C6EE00100A |
| [12] |
A. Mendoza-Garcia, D. Su, S. Sun, Nanoscale 8 (2016) 3244-3247. DOI:10.1039/C5NR08763E |
| [13] |
Y. Liu, C. Xiao, M. Lyu, et al., Angew. Chem. 127 (2015) 11383-11387. DOI:10.1002/ange.201505320 |
| [14] |
J.S. Chen, J. Ren, M. Shalom, T. Fellinger, M. Antonietti, ACS Appl. Mater. Interfaces 8 (2016) 5509-5516. DOI:10.1021/acsami.5b10099 |
| [15] |
T.Y. Ma, J.L. Cao, M. Jaroniec, S.Z. Qiao, Angew. Chem. Int. Ed. 55 (2016) 1138-1142. DOI:10.1002/anie.201509758 |
| [16] |
P. Chen, K. Xu, Z. Fang, et al., Angew. Chem. 127 (2015) 14923-14927. DOI:10.1002/ange.201506480 |
| [17] |
Y. Liu, H. Cheng, M. Lyu, et al., J. Am. Chem. Soc. 136 (2014) 15670-15675. DOI:10.1021/ja5085157 |
| [18] |
A.T. Swesi, J. Masud, M. Nath, Energy Environ. Sci. 9 (2016) 1771-1782. DOI:10.1039/C5EE02463C |
| [19] |
A. Grimaud, K.J. May, C.E. Carlton, et al., Nat. Commun. 4 (2013) 2439. DOI:10.1038/ncomms3439 |
| [20] |
S. Chen, J. Duan, M. Jaroniec, S. Z. Qiao, Adv. Mater. 26 (2014) 2925-2930. DOI:10.1002/adma.v26.18 |
| [21] |
J. Zhang, Z. Zhao, Z. Xia, L. Dai, Nat. Nanotechnol. 10 (2015) 444. DOI:10.1038/nnano.2015.48 |
| [22] |
J. Yang, G. Zhu, Y. Liu, et al., Adv. Funct. Mater. 26 (2016) 4712-4721. DOI:10.1002/adfm.v26.26 |
| [23] |
J. Jiang, C. Yan, X. Zhao, et al., Green Chem. 19 (2017) 3023-3031. DOI:10.1039/C7GC01012E |
| [24] |
P. Cai, J. Huang, J. Chen, Z. Wen, Angew. Chem. Int. Ed. 56 (2017) 4858-4861. DOI:10.1002/anie.201701280 |
| [25] |
B. Dong, X. Zhao, G.Q. Han, et al., J. Mater. Chem. A 4 (2016) 13499-13508. DOI:10.1039/C6TA03177C |
| [26] |
S. Dou, L. Tao, J. Huo, S. Wang, L. Dai, Energy Environ. Sci. 9 (2016) 1320-1326. DOI:10.1039/C6EE00054A |
| [27] |
P. Ganesan, M. Prabu, J. Sanetuntikul, S. Shanmugam, ACS Catal. 5 (2015) 3625-3637. DOI:10.1021/acscatal.5b00154 |
| [28] |
M. Shen, C. Ruan, Y. Chen, et al., ACS Appl. Mater. Inter. 7 (2015) 1207-1218. DOI:10.1021/am507033x |
| [29] |
X. Long, J. Li, S. Xiao, et al., Angew. Chem. 126 (2014) 7714-7718. DOI:10.1002/ange.201402822 |
| [30] |
T. Xia, J. Wang, K. Jiang, et al., Chin. Chem. Lett. 29 (2018) 861-864. DOI:10.1016/j.cclet.2017.10.038 |
| [31] |
S. Zheng, H. Xue, H. Pang, Coord. Chem. Rev. 373 (2018) 2-21. DOI:10.1016/j.ccr.2017.07.002 |
| [32] |
S. Zheng, X. Li, B. Yan, et al., Adv. Energy Mater. 7 (2017) 1602733. DOI:10.1002/aenm.201602733 |
| [33] |
Y. Wang, L. Tao, Z. Xiao, R. Chen, Z. Jiang, S. Wang, Adv. Funct. Mater. 28 (2018) 1705356. DOI:10.1002/adfm.v28.11 |
| [34] |
H. Liang, L. Li, F. Meng, L. Dang, J. Zhuo, Chem. Mater. 27 (2015) 5702-5711. DOI:10.1021/acs.chemmater.5b02177 |
| [35] |
B. Chen, R. Li, G. Ma, et al., Nanoscale 7 (2015) 20674-20684. DOI:10.1039/C5NR07429K |
| [36] |
A. Aijaz, J. Masa, C. Rösler, et al., Angew. Chem. Int. Ed. 55 (2016) 4087-4091. DOI:10.1002/anie.201509382 |
| [37] |
C. Sun, Q. Dong, J. Yang, et al., Nano Res. 9 (2016) 2234-2243. DOI:10.1007/s12274-016-1110-1 |
| [38] |
Z. Zhang, J. Hao, W. Yang, J. Tang, ChemCatChem 7 (2015) 1920-1925. DOI:10.1002/cctc.v7.13 |
| [39] |
J. Shao, Z. Wan, H. Liu, et al., J. Mater. Chem. A 2 (2014) 12194-12200. DOI:10.1039/C4TA01966K |
| [40] |
W. Zeng, G. Zhang, X. Wu, et al., J. Mater. Chem. A 3 (2015) 24033-24040. DOI:10.1039/C5TA05934H |
| [41] |
Y. Hou, J. Li, Z. Wen, et al., Nano Energy 12 (2015) 1-8. DOI:10.1016/j.nanoen.2014.11.043 |
| [42] |
S. Liu, J. Wang, J. Wang, et al., CrystEngComm 16 (2014) 814-819. DOI:10.1039/C3CE41915K |
| [43] |
J. Zhu, Z. Ren, S. Du, et al., Nano Res. 10 (2017) 1819-1831. DOI:10.1007/s12274-017-1511-9 |
| [44] |
C. Wu, Y. Zhang, D. Dong, H. Xie, J. Li, Nanoscale 9 (2017) 12432-12440. DOI:10.1039/C7NR03950F |
| [45] |
M. Chauhan, K.P. Reddy, C.S. Gopinath, S. Deka, ACS Catal. 7 (2017) 5871-5879. DOI:10.1021/acscatal.7b01831 |
| [46] |
Y. Kim, J.B. Goodenough, J. Phys. Chem. C 112 (2008) 15060-15064. DOI:10.1021/jp8038847 |
| [47] |
N. Jiang, L. Bogoev, M. Popova, et al., J. Mater. Chem. A 2 (2014) 19407-19414. DOI:10.1039/C4TA04339A |