 2019, Vol. 30
2019, Vol. 30 


b School of Chemistry and Chemical Engineering, Shandong University, Ji'nan 250100, China;
c Fujian Key Laboratory of Photoelectric Functional Materials, College of Materials Science and Engineering, Huaqiao University, Xiamen 361021, China
The splitting of water via artificial photosynthesis (AP) is currently attracting immense attention from chemists because of the significance of the conversion of solar energy into chemically stored energy in the form of clean fuel hydrogen (H2) [1, 2]. In general, an efficient AP system for photochemical reduction of protons to H2, contains a photosensitizer (PS) for harvesting light energy and transforming it into electrochemical energy, and a proton-reducing catalyst as electron acceptor and catalyzing the proton reduction, and a sacrificial electron donor to allow the reductive half-reaction to be studied separately. As light harvesters desirable for AP systems, noble-metal-based complexes are most widely employed. However, these complexes are limited by the high cost and low supply of noble metals. As an alternative to noble-metal complexes, organic dyes show potential candidates as PSs due to their less extensive cost and readily adjustable structure and chemical properties [3, 4]. Among a few organic dyes, BODIPY (boron dipyrromethene, IUPAC name: 4, 4-difluoro-4-bora-3a, 4a-diaza-s-indacene) compounds have been tested as cheap alternative light harvesters for light-driven proton reduction due to their high extinction coefficients and possibilities for more sophisticated PSs design [5-13].
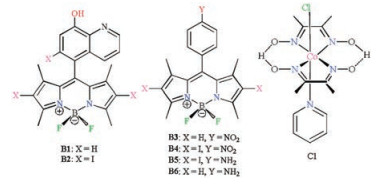
However, similar to xanthenes dyes [14], these reported BODIPY compounds as PSs for light-driven hydrogen evolution in combination with the catalyst [CoⅢ(dmgH)2PyCl] (dmgH = dimethylglyoximate, and py= pyridine) and electron donors triethanolamine (TEOA) or triethylamine (TEA) are usually performed in the basic conditions [5-9, 15-17]. We wonder whether BODIPY chromophores can be carried out in the acidic solutions only by modifying their substitution patterns. To this end, in the present work, BODIPY dyes having 8-hydroxyl-quinoline or phenylamine moiety at the meso-position on the BODIPY core were prepared. The detailed molecular structures of dyes are displayed in Fig. 1. These BODIPY dyes were tested for investigating the structure-and-property relationship between the BODIPY structure and activity of visible-light-driven H2 production as well as solution pH.

|
Download:
|
| Fig. 1. Structures of BODIPY PSs (B1–B6) and catalyst C1 employed in this study. | |
There BODIPY dyes were synthesized according to the previous literature procedures [18-23]. The photophysical properties of the BODIPYs were firstly measured in deaerated dry acetontrile (MeCN) via UV-vis absorption and steady-state fluorescence spectroscopy (Fig. S1 in Supporting information), and the corresponding data are summarized in Table 1. As shown in Fig. S1a, the absorptionspectrum of B1 withagroup of8-hydroxyl-quinoline-5-yl at the meso-position on the BODIPY core is of comparable shape as those of described BODIPY dyes [5-10], having a strong S0→S1(π→π*) transition centered at 501 nm and a weaker, broad S0→S2 band centered at 370 nm. Upon introduction ofiodine atoms at the pyrrole carbons ofthe BODIPYcore in B2 showed S0→S1 transition which experienced a red-shift of ~36 nm compared to that of B1. The emission spectra of B1 and B2 were mirror images of the absorbance spectra with small Stokes' shifts in the range of 760-580cm-1, implying there is little change in geometry or polarity between ground and excited states. The fluorescence quantum yield (Φf1) of B1 in deoxygenated acetonitrile solution at room temperature was found to be ~0.30. In comparison, iodinated B2 showed substantial reduction of fluorescence quantum yield (Φf1 = 0.007). A significant decrease of Φf1 indicates that the presence of heavy atom Ⅰ will facilitate an efficient intersystem crossing efficiency from the lowest singlet excited state to the triplet states, resulting in population of the longer lived triplet excited states from which electron transfer occurs. As shown in Fig. S1b, replacement of the 8-hydroxyl-quinoline substituent with a phenylamine moiety on the meso place on the BODIPY core does not significantlyalter the absorption and emission maxima of the dyes. Similarly, the addition of iodo atoms results in absorption and emission energies that are shifted to lower energy. Notably, iodinated B5 augmented the absorption compared with iodinated B2 (ε = 86800Lmol-1cm-1 for B5 vs. ε = 79800Lmol-1cm-1 for B2).
|
|
Table 1 Electrochemical, absorption and emission data.a, b |

{kind=link}
The oxidation and reduction potentials of BODIPYs (B1-B2 and B5-B6) were further studied by cyclic voltammograms in dry MeCN using tetraburylammonium hexafluorophosphate (TBAP) as a supporting electrolyte, and the corresponding electrochemical data are also gathered in Table 1. As observed with other BODIPY PSs [5, 6], within the electrochemical window of solvent MeCN, all these BODIPYs exhibited one-electron oxidation and reduction. Compared to non-iodinating BODIPYs (B1 and B6), iodinating BODIPYs (B2 and B5) with two iodo atoms on the 2, 6-positions, exhibited less negative reduction and more positive oxidation potentials, indicating that iodinated compounds (B2 and B5) are easier to get one electron to be reduced than the corresponding counterparts (B1 and B6). This suggests the electron-deficient nature of iodo-substituted BODIPYs due to the presence of iodine atoms at the core of the pyrrole [7]. In addition, the electron-deficient nature of iodo-substituted BODIPYs will be also easier to form radical anions in the presence of appropriate electron donors during the course of light-driven proton reduction.
In most of the hydrogen-producing systems reported using C1 as the catalyst and BODIPYor xanthene dyes as the PSs, irreversible electron donors such as triethanolamine (TEOA) or triethylamine (TEA) were frequently used. However, when TEOA or TEA was initially chosen as electron donors with B2 serving as the PS and C1 as the catalyst, no H2 gas was detected by GC analysis after 6h of visible-light irradiation. Despite the negative results regarding H2 generation in the neutral and basic aqueous solution, one electron reduced BODIPY species obtained from the excited state ofBODIPY (BODIPY*) and electron donor is thermodynamically able to reduce the catalyst C1 (ΔG = E(BODIPY*-/BODIPY) — E(CoⅢ/CoⅡ) = —0.65 eV), which encourages us to explore the three-component system in the acidic conditions. Ascorbic acid (H2A) is widely used as an electron donor in acidic photocatalytic H2 evolution system.
In an initial experiment, visible light (λ > 420 nm) irradiation of 10mL of aqueous solution (VMeCN:VH2O = 1:1) containing B2 (1.0×10—4mol/L), C1 (2.5×10—3mol/L) and H2A (0.1mol/L) at pH 4, leads to hydrogen production (Fig. 2a). Control experiments suggested that the system is inactive in the absence of any of the three components (catalyst C1, PS B2 or sacrificial donor H2A). The formation of Co colloids as possible catalytically active species in the course of the photocatalysis was ruled out on the basis of Hg-poisoning experiments. It should be noted that when using H2A as electron donor, the net reaction being driven photochemically can be expressed by the following equation: H2A→A+H2. The formation of A and H2 from H2A has been determined to be thermodynamically unfavorable by 0.41 V at pH 4. Thus, photocatalytic H2 generation from H2A represents a photochemically driven upconversion of ~20kcal/mol. The visible light-driven H2 production was dependent on the solution pH. A maximal rate for H2 evolution was achieved at pH 5 (Fig. 2b), while lower amountsof H2 were obtained at either lower or higher pH values, similar to that in other systems [24].This pH dependenteffect maybe related to several factors, including the protonation behavior of the PS, and the equilibrium of H2A ↔ H+ + HA- (pKa = 4.03 and 3.90 for H2A and HA-, respectively).

|
Download:
|
| Fig. 2. (a) Photoinduced H2 evolution using H2A, TEOA or TEA as sacrificial electron donor with C1 (2.5×10~3 mol/L) and B2 (1.0 × 10~4 mol/L) in MeCN/H2O (1:1, v/v) after 6h irradiation; (b) Effect of solution pH on the photocatalytic H2 production from a system composed of C1, B2 and H2A in MeCN/H2O 1:1. | |
{kind=link}
In order to investigate and compare the ability of B1-B2 and B5-B6 acting as PSs for H2 evolution, a series of photolysis experiments were performed under the same reaction conditions in the presence of C1 (2.5×10-3mol/L), PS (1.0 × 10-4mol/L), and H2A (0.1 mol/L) with continuous visible-light irradiation (λ > 420nm). The optimal pH of the reaction system was kept at optimal value of 5. Fig. 3 shows the time dependence of H2 production in this catalytic system for the different BODIPY PSs. The plot reveals: (1) No observable amount of H2 production was detected by GC for B1 and B5. As reported in previous studies [7], compared with iodinated BODIPY dyes noniodinated chromophores B1 and B2 lack any appreciable hydrogen evolution under identical conditions due to the absence of the internal heavy atom effect and the long-lived photoexcited triplet state. The reason is that the diffusion-controlled bimolecular collision frequency in fluid solution is 109-1010Lmol-1s-1. As a consequence, the Stern-Volmer quenching constant (Ksv = kq × τ0, where kq is the bimolecular quenching constant and τ0 is the lifetime of the excited state of the PS (inns)) will be on the scale of 1-10L/mol, which is too small to induce any efficient intermolecular electron transfer. (2) B2 and B6 show significant amounts of hydrogen. Iodination of BODIPY PSs (B2 and B5) increase the rate of intersystem crossing from the initially excited 1ππ* state via internal heavy atom effect, thus generating greater amounts of 3ππ* state. The excited triplet state tends to have a longer lifetime (in ms to ms) than the corresponding excited singlet state (inns), thereby allowing diffusion-controlled bimolecular interactions to occur more substantially. Although the triplet lifetimes for B5 and B2 were not measured due to our instrumental limitation, the analog of B5, not having the -NH2 substituent, has a reported triplet excited-state lifetime of 57.1 ms in MeCN, which is > 30000-fold longer than the corresponding singlet excited state. Furthermore, an additional iodide source such KI showed no enhancement of H2 production compared to the iodine-free system of C1/B1 or C1/B6, thus exclude the role of external heavy atom effect. (3) B5 and B2 gave 930 and 622 μmol ofH2 evolution after 6 h irradiation, respectively, corresponding toTONs of 93 and 62 with respective to the PS. Generally, multicomponent photocatalytic systems with different dyes always exhibit different photocatalytic H2 evolution behavior due to their different light-absorbing properties and long-lived 3ππ* states generation capacities. When compared with B2 having one 8-hydroxyl-quinoline moiety, B5 with one phenyl-amine substituent shared a similar absorption and emission profile, but augmented the absorption (ε = 86800 L mol —1 cm—1 for B5 vs. ε = 79800 L mol —1 cm—1 for B2).

|
Download:
|
| Fig. 3. Time courses of photocatalytic H2 evolution in PS/C1 systems with different BODIPY dyes (B1-B2 and B5-B6). Conditions: CH3CN-H2O (1:1, v/v) at pH 5 containing a BODIPY PS (1.0 × 10" 4 mol/L), C1 (2.5 × 10" 3 mol/L) and H2A (0.1 mol/L) under visible light irradiation (300 W Xe lamp, λ > 420 nm). | |
{kind=link}
After approximately 6 h of visible-light irradiation, the activity of H2 evolution in the system of C1 /B5/H2A ceased, indicating that at least one of the system components had been consumed. Addition of one equivalent of B5 to the catalytic systems after 6 h of irradiation partially restores the H2 evolution activity (Fig. S2a, Supporting information), implying that the system is mainly limited by the decomposition of the PS and the catalyst. The photodegradation of the PS and the catalyst is also evidence by comparison of the following absorption spectra recorded during the photolysis processes. The absorption spectra of the irradiated B5 solution show that 40 min irradiation did not produce fading of B5 (Fig. S2b), indicating the high photostability of iodinated BODIPY [21]. As shown in Fig. S2c, in the presence of only sacrificial donor, B5 bleaches very rapidly as a result of the instability of the formed radical anion B5·— upon reductive quenching electron transfer between excited state of B5 and H2A.The peak position was shifted ~510nm due to the cleavage of C-I bond. It has been documented that the halogen groups can be easily photobleached through reductive quenching [7]. The absorption spectra of the irradiated B5 + C1 + H2A solution display that the decomposition of B5 becomes much slower (Fig. S2d) by the addition of C1 as the electron acceptor, which is attributed to the fact that the radical anion B5·— may transfer an electron to C1 and regenerates B5.
As mentioned in the context, an interesting feature of the photogeneration of H2 systems of C1/B2 and C1/B5 is that they keep active not in the basic solutions but in the acidic conditions. A possible, although still ambiguous, speculation may be rationalized from the structural analysis of BODIPYs. Taking the PS B5 for example, to minimize unfavorable steric clashes between the ortho-hydrogen atoms and the vicinal methyl groups, the orthogonal arrangement of the phenylamino and dipyrromethene fragments is anticipated in the B5. Thus, the rapid intramolecular electron transfer across the orthogonal structure will be favorable in the B5. In other words, the content of competition exists between intramolecular and intermolecular electron transfer in the basic and acidic conditions (Scheme S1 in Supporting information). Specifically, in the basic conditions, the phenylamino unit may serve as electron transfer donor, rapidly quenching the singlet or triplet BODIPY* state intramolecularly and preventing electron transfer to the catalyst C1. The control experiment thus introduced an alternative reaction path forward electron transfer from phenylamino group to the excited state of BODIPY* followed by back electron transfer to ground state that prevents H2 production. In contrast, once the phenylamino unit is protonated in acidic solutions, the path of intramolecular electron transfer is restricted and intermolecular electron transfer from dipyrrome-thene unit to the catalyst C1 is favorable. As a consequence, the activityofhydrogen production is restored. It should be noted that the current mechanism is only preliminary speculation on the basis of structure of BODIPYs. Further work including transient absorption spectroscopy and theoretical calculations should needed to shed light on the detailed mechanistic process.
To conclude, a series ofBODIPY dyes with 8-hydroxyl-quinoline or phenylamine moiety at the meso-position on the BODIPY core were tested as PSs in the three-component homogeneous photocatalytic system containing the cobaloxime catalyst C1 and H2A as the electron donor in aqueous organic media. Only systems containing iodinated BODIPY chromophores were found to be active for H2 production in the acidic aqueous solutions. This is the first time for the BODIPYs as homogeneous hydrogen-generating PSs used in the acidic aqueous solutions. The results underscore that the chemical modification of PSs can be performed, thus allowing for the transformation of acid and base conditions for the light-driven hydrogen production.
AcknowledgmentsWe are grateful to the National Natural Science Foundation of China (Nos. 216410H, 21571H5 and 2170H33), the Program for New Century Excellent Talents in Fujian Province University and the Fujian Key Laboratory of Functional Materials and Applications (No. fma2017107), the Natural Science Foundation of Shandong Province (No. JQ201803), Young Scholars Program of Shandong University (No. 2015WLJH24), and the Fundamental Research Funds of Shandong University (No. 104.205.2.5) for financial support of this work.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/jxclet.2018.05.003.
| [1] |
Z.J. Han, R. Eisenberg, Acc. Chem. Res. 47 (2014) 2537-2544. DOI:10.1021/ar5001605 |
| [2] |
P.W. Du, R. Eisenberg, Energy Environ. Sci. 5 (2012) 6012-6021. DOI:10.1039/c2ee03250c |
| [3] |
G.G. Luo, H. Lu, Y.H. Wang, J. Dong, Y. Zhao, R.B. Wu, DyesPigm. 134 (2016) 498-505. |
| [4] |
A. Xiao, H. Lu, Y. Zhao, G.G. Luo, Acta Phys.-Chim. Sin. 32 (2016) 2968-2975. |
| [5] |
G.G. Luo, K. Fang, J.H. Wu, et al., Phys. Chem. Chem. Phys. 16 (2014) 23884-23894. DOI:10.1039/C4CP03343D |
| [6] |
J. Bartelmess, A.J. Francis, K.A.E.I. Roz, et al., Inorg. Chem. 53 (2014) 4527-4534. DOI:10.1021/ic500218q |
| [7] |
G.G. Luo, H. Lu, X.L. Zhang, et al., Phys. Chem. Chem. Phys. 17 (2015) 9716-9729. DOI:10.1039/C5CP00732A |
| [8] |
G.G. Luo, K. Fang, J.H. Wu, et al., Chem. Commun. 51 (2015) 12361-12364. DOI:10.1039/C5CC03897A |
| [9] |
R.P. Sabatini, B. Lindley, T.M. McCormick, et al., J. Phys. Chem. B 120 (2016) 527-534. |
| [10] |
L. Dura, J. Ahrens, M. Pohl, et al., Chem.-Eur. J. 21 (2015) 13549-13552. DOI:10.1002/chem.201501637 |
| [11] |
B. Zheng, R.P. Sabatini, W.F. Fu, et al., Proc. Natl. Acad. Sci. U. S. A. 112 (2015) E3987-E3996. DOI:10.1073/pnas.1509310112 |
| [12] |
A. Prlj, L. Vannay, C. Corminboeuf, Helv. Chim. Acta 100 (2017) e1700093. DOI:10.1002/hlca.v100.6 |
| [13] |
P. Farrás, A.C. Benniston, Tetrahedron. Lett. 55 (2014) 7011-7014. DOI:10.1016/j.tetlet.2014.10.119 |
| [14] |
T. Lazarides, T. McCormick, P.W. Du, et al., J. Am. Chem. Soc. 131 (2009) 9192-9194. DOI:10.1021/ja903044n |
| [15] |
G.G. Luo, Y.H. Wang, J.H. Wu, et al., Chem. Commun. 53 (2017) 7007-7010. DOI:10.1039/C7CC01942D |
| [16] |
Y. Zhao, Y.H. Wang, Q.Y. Wu, et al., Chin. J. Catal. 39 (2018) 517-526. DOI:10.1016/S1872-2067(17)62940-1 |
| [17] |
G.G. Luo, X.C. Li, J.H. Wang, ChemistrySelect 3 (2016) 425-429. |
| [18] |
Y.H. Zhong, L.P. Si, H.S. He, et al., Dalton Trans. 40 (2011) 11389-11395. DOI:10.1039/c1dt11137j |
| [19] |
H. He, L. Si, Y. Zhong, et al., Chem. Commun. 48 (2012) 1886-1888. DOI:10.1039/c2cc17037j |
| [20] |
Z.H. Pan, G.G. Luo, J.W. Zhou, et al., Dalton Trans. 43 (2014) 8499-8507. DOI:10.1039/C4DT00395K |
| [21] |
T. Yogo, Y. Urano, Y. Ishitsuka, et al., J. Am. Chem. Soc. 127 (2005) 12162-12163. DOI:10.1021/ja0528533 |
| [22] |
T. Yogo, Y. Urano, A. Mizushima, et al., Proc. Natl. Acad. Sci. U. S. A. 105 (2008) 28-32. DOI:10.1073/pnas.0611717105 |
| [23] |
Z.H. Pan, J.W. Zhou, G.G. Luo, et al., Phys. Chem. Chem. Phys. 16 (2014) 16290-16301. DOI:10.1039/C4CP02151G |
| [24] |
K.A. Brown, S. Dayal, X. Ai, et al., J. Am. Chem. Soc. 132 (2010) 9672-9680. DOI:10.1021/ja101031r |