 2019, Vol. 30
2019, Vol. 30 


b CAS Key Laboratory of Organic Solids, CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
Organic solar cells (OSCs) are regarded as a potential photovoltaic technology to convert sunlight into electricity due to theiradvantages of flexibility, light weight, large-area capability, and easy fabrication [1-6]. Typically, the active layer of an organic solar cell is a bulk-or bilayer-heterojunction consisting of two components, an electron-donating and an electron-accepting material [7-12]. Owing to the design of new active materials and optimization of processing conditions, great advances have achieved in the power conversion efficiencies (PCEs) of OSCs [13-29]. Over the past twenty years, fullerene derivatives (e.g., PCBM and ICBA) were dominantly used as electron acceptors due to their superior electron affinity and high electron mobility [30-34]. However, these acceptor materials have some intrinsic limitations, including weak optical absorption, untunable energy levels, and high costs [35, 36].
In recent years, non-fullerene small-molecule acceptors have attracted increasing attention due to their strong and broad absorption and highly tunable electronic energy levels [35, 37-39]. A large number of electron acceptors were developed on the base of fused or non-fused aromatics. Most strikingly, the indaceno-dithiophene (IDT)-based A-π-A electron acceptors, such as ITIC and IEIC [40, 41], have achieved remarkable breakthrough in organic photovoltaics (OPVs). To date, the most efficient binary non-fullerene OPV devices using IEIC and ITIC as electron acceptor have gained PCEs up to 10% and 13.1% [29, 42], respectively. The PCEs can be further improved by fabricating ternary or tandem OPV devices [42-45].
Relative to aromatic systems, anti-aromatics are characteristic of smaller energy gap and deeper electron affinity [46-53], which is beneficial for broadening optical absorption and increasing electron-accepting ability. For instance, quinoidal indenofluorenes (IFs) based on the s-indacene anti-aromatic framework exhibit electron affinity higher than PCBM [54-56]. What is more, IFs possess broad absorption with the absorption edge extending to the near infrared [57-60]. In addition, good and ambipolar charge transport properties have been found in the IFs based field-effect transistors [61-63]. Despiteofthese desirableproperties, veryfew work was reported on the OPVs based on "anti-aromatic" electron acceptors [64-66].
In this contribution, we have designed a series of A-π-A small-molecule electron acceptors, which consist of anti-aromatic s-indacene based π-bridges and the electron-withdrawing groups of 2-(3-oxo-2, 3-dihydroinden-1-ylidene) malononitrile (INCN). In order to assess the potential of the "anti-aromatic" acceptors in OPVs, we have calculated their electronic, optical, and charge transport properties in comparison with the corresponding "aromatic" electron acceptors by (time-dependent) density functional theory (the computational details see Supporting information).
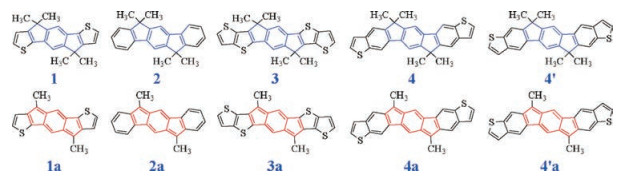
The chemical structures of the anti-aromatic and aromatic fused π-units are shown in Scheme 1. All the aromatic polycyclic π-units (1-4 and 4´) contain a core of 1, 5-dihydro-s-indacene (in blue). When the core is fused with two thiophene or benzene moieties, we get units 1 (indacenodithiophene, IDT) or 2 (indacenodibenzene, IDB). Further fusion of two thiophenes onto units 1 or 2 will return units 3 (indacenodithienothiophene, IDTT) or 4/4´ (indacenodibenzothiophene). Isomers 4 and 4´ are different in sulfur orientations of the fused thiophenes. Units 1 (IDT) and 3 (IDTT) have been reported as the backbone components ofIEIC and ITIC, respectively. Correspondingly, the anti-aromatic polycyclic π-units (1a-4a and 4´a) are designed by replacing the aromatic 1, 5-dihydro-s-indacene core with the anti-aromatic s-indacene (in red), as shown in Scheme 1. The bond lengths of the optimized geometries for the π-units are shown in Fig. S1 (Supporting information). The bond length alternations are significant in the s-indacene core. Overall, the C—C bonds of benzene are longer in the anti-aromatic indacene than in the aromatic indacene; in contrast, the C—C bonds of cyclopentene are much shorter in the antiaromatic indacene than the aromatic indacene (Fig. S2 in Supporting information).

|
Download:
|
| Scheme 1. Chemical structures of the aromatic fused π-units (top) and corresponding anti-aromatic fused π-units (bottom). | |
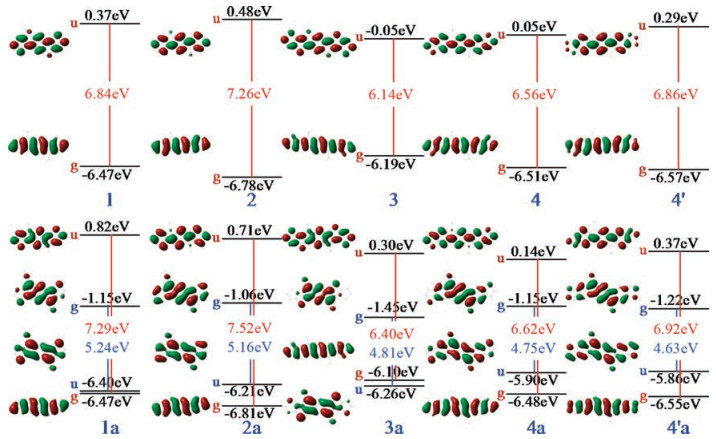
The calculated frontier orbital energy diagram and pictorial representation of these π-units are displayed in Fig. 1. The HOMOs (centrosymmetric, g) and LUMOs (anti-centrosymmetric, u) of all the aromatic π-units are delocalized over the whole backbones while the LUMO of 4´ shows relatively weak distribution on the lateral atoms of the fused thiophenes. When replacing thiophene with benzene fused onto the aromatic indencene core, the HOMO level will descend appreciably by 0.31 eV but the LUMO level will ascend slightly by 0.11 eV, leading to an increment of 0.42 eV in the LUMO-HOMO energy gap (Egap) from 1 to 2. Similar energy changes are found from 3 to 4, but more apparent from 3 to 4´; particularly, the increase of the LUMO energy from 4 to 4´ even reaches 0.24 eV due to the relatively weakened delocalization. As expected, extension of the π conjugation from 1 (2) to 3 (4/4´) results in lower LUMO and higher HOMO levels and smaller gaps.

|
Download:
|
| Fig. 1. Energy diagram and pictorial representation for the frontier orbitals of the aromatic 1-4/4´ and anti-aromatic 1a-4a/4´a π-units. | |
Interestingly, the anti-aromatic π-units 1a-4a and 4´a have two pairs of important frontier molecular orbitals. One pair of the frontier orbitals correspond to the HOMO and LUMO of the aromatic units 1-4 and 4´, which we name as "aromatic" orbitals. Compared with 1-4 and 4´, the "aromatic" unoccupied orbital energies are significantlyup-shifted while the "aromatic" occupied orbital energies are hardly changed for 1a-4a and 4´a. The other pair of frontier orbitals of 1a-4a and 4´a originate from the antiaromatic s-indacene, which are named as "anti-aromatic" orbitals. These "anti-aromatic" orbitals display a quinoidal π-conjugation character dictated by the significant bond length alternation in the s-indacene core. The "anti-aromatic" unoccupied orbital levels (centrosymmetric, g) are lower than the "aromatic" unoccupied orbitals by ca. 1.3-2.0 eV. As a result, the LUMO energies are substantially (at least 1.2 eV) decreased for the anti-aromatic units relative to the aromatic units, indicating that introducing antiaromatic structures can enhance electron-accepting abilities of the π-units. In contrast, the energy differences are much smaller between the "anti-aromatic" and "aromatic" occupied orbitals, ca. 0.6-0.7 eV for 2a, 4a and 4´a and even only 0.07 eV and 0.16 eV for 1a and 3a, respectively. Both the occupied and unoccupied "antiaromatic" orbitals are mainly localized on the s-indacene core for 1a and 3a, but more extended for 2a, 4a and 4´a. Also, the electron distributions of the "anti-aromatic" orbitals can be modulated by the fused thiophene orientations; higher electron densities are found on the peripheral atoms of the fused thiophenes for 4´a relative to 4a.
The UV-vis absorption spectra of all the π-units are shown in Fig. S3 (Supporting information), and the electronic transition properties of the lowest excited singlet states (S1) are summarized in Table S1 (Supporting information). For the aromatic π-units, only a single absorption peak appears in the UV region, corresponding to the S1 excitation dominated by the HOMO→LUMO transition. Consistent with the ordering of Egap, the wavelength at the maximum absorption is red-shifted as follows 2 < 1 and 4´ < 4 < 3. For the anti-aromatic π-units, the absorption from the transition between the "aromatic" occupied and unoccupied frontier orbitals is significantly blue-shifted and much stronger due to larger energy gap with respect to the aromatic π-units. Besides, an additional weaker absorption appears in the visible region owing to the lower-energy transition between the "anti-aromatic" occupied and unoccupied frontier orbitals. It should be noticed that the S1 excitation for the anti-aromatic units is dominated by the transition between the "aromatic" occupied orbital and "anti-aromatic" unoccupied orbital and is symmetry forbidden. To summarize, both isomerization and anti-aromatization have important impact on the electronic and optical properties of the π-units.
The chemical structures of all the A-π-A acceptor molecules are shown in Scheme 2. For the acceptors containing the short fused-ring units (A1/A1a, A2/A2a, and A2´/A2´a), the π-units are extended by linking two additional thiophene moieties. The structure difference between A2 and A2´ or A2a and A2´a is the linking positions on the fused-ring units. Most of those molecules show a completely flat backbone (Figs. S4 and S5 in Supporting information). In the case of A2/A2´ and A2a/A2´a, the fused-ring core and the thiophene moieties exhibit a twisted angle of 22.4°/ 26.6° for the aromatic molecules and 22.7°/21.3° for the anti-aromatic molecules.

|
Download:
|
| Scheme 2. Chemical structures of the "aromatic" and "anti-aromatic" acceptors. | |
Fig. 2 and Fig. S6 (Supporting information) show the frontier molecular orbitals and corresponding energies for the acceptors based on the short and long fused units, respectively. As seen from Fig. S7 (Supporting information), the HOMO/LUMO energies of INCN (-9.00eV/-1.65eV) are substantially lower than the "aromatic" frontier levels of the π-units, especially for the HOMO. As a result, the HOMO (g-symmetry) of the "aromatic" acceptors is determined by the π-bridges. Consistent with the trends in the π-units, the HOMO levels of the acceptors are successively decreased as follows: A1 > A2 > A2´ and A3 > A4 > A4´ (Fig. 3). In contrast, the LUMO (u-symmetry) and LUMO +1 (g-symmetry) of the "aromatic" acceptors are dominated by the two terminal INCN units. Compared with A1 (IEIC) or A3 (ITIC), the LUMO electron densities are more localized on the INCN groups for A2/A2´ or A4/ A4´, in particular for A2´ and A4´. Because of the reduced electronic delocalization, the LUMO level is gradually up-shifted by nearly 0.3 eV (A1 < A2 < A2´ and A3 < A4 < A4´). Relatively, the LUMO +1 electron densities and energy levels are slightly changed. Thus, the LUMO and LUMO +1 energies are almost degenerate for A2´ and A4´. The increased LUMO levels are beneficial for achieving high open circuit voltages.

|
Download:
|
| Fig. 2. Frontier orbital energies and pictorial representation for the A-π-A acceptors based on (a) the aromatic (Al, A2 and A2´) and (b) the anti-aromatic s-indacene cores (A1a, A2a, and A2´a). | |

|
Download:
|
| Fig. 3. Frontier orbital energies of the A-π-A acceptors based on (a) the aromatic and (b) the anti-aromatic s-indacene cores (red: u-symmetry, black: g-symmetry). | |
Compared to the "aromatic" acceptors, the "anti-aromatic" acceptors have one more anti-centrosymmetric occupied frontier orbital and one more centrosymmetric unoccupied frontier orbital (Fig. 2 and Fig. S6), which are brought about by the anti-aromaticity of the s-indacene core. The "aromatic" occupied frontier orbitals of the "anti-aromatic" acceptors are similar to the HOMOs of the "aromatic" acceptors; except A4a, the orbital energies are slightly higher (Fig. 3). The "anti-aromatic" occupied frontier orbitals are localized on the fused-ring cores for A1a/A2a and A3a/A4a while extended to the INCN groups for A2´a and A4´a due to effective couplings of the fused-ring units with the rest moieties caused by the large electron densities on the linking atoms; the energy change trend appears to be opposite to the "aromatic" occupied frontier orbitals. Consequently, the HOMO is "aromatic" for A1a and A3a but "anti-aromatic" for A2a/A2´a and A4a/A4´a. The anti-centrosymmetric unoccupied frontier orbitals are similar to those of the "aromatic" acceptors while their energies are a bit lower. In the case of the centrosymmetric unoccupied frontier orbitals, the "anti-aromatic" orbitals are concentrated on the s-indacene core and isolated from the INCN-dominant ones for A1a and A3a; on the contrary, the "anti-aromatic" and INCN-dominant components are hybridized for A2a/A2´a and A4a/A4´a. Especially for A2´a and A4´a, large energy splitting (> 0.6 eV) is found between these two centrosym-metric unoccupied orbitals due to strong electronic interaction; hence the LUMO becomes to be of g-symmetry. The remarkable changes in the frontier molecular levels are expected to have profound influence on the optical absorption properties of the electron acceptors.
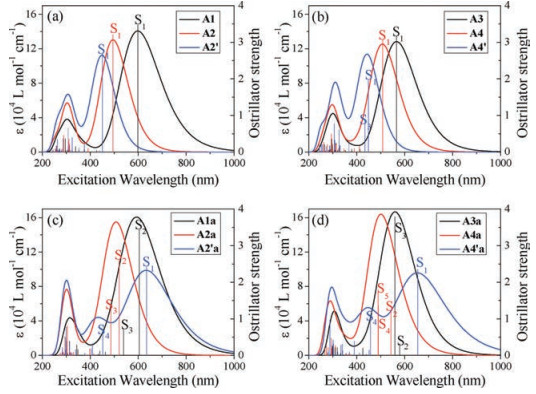
The UV-vis absorption spectra of all the A-π-A acceptors are shown in Fig. 4 and corresponding excitation properties are summarized in Table 1. All the acceptors have similar and moderate absorption in the UV range of 250-350 nm, which is composed of many high-energy excitations. Here we focus on the absorption spectra at the long wavelengths above 350 nm. As seen from Fig. 4a, the "aromatic" acceptors Al, A2, and A2´ exhibit a single absorption peak, which is attributed to the S1 excitation. From A1 to A2 and A2´, the contribution to the S1 excitation is decreased for the HOMO→LUMO transition while increased for the other higher-energy transitions (Table 1); along with the enlarged energy gap (Fig. 3), the absorption peak is then obviously blue-shifted with decreased intensity. Similar trend in the absorption is found from A3 to A4 and A4´. We noticed that the absorption of A4´ arises from both S1 and S3 excitations, which have close energies and consist of the same main transitions with different percentages.
|
|
Table 1 Excitation wavelengths (λ, nm), oscillator strengths (f), and main electronic transitions and corresponding weights for the excited states of the A-π-A acceptors. |
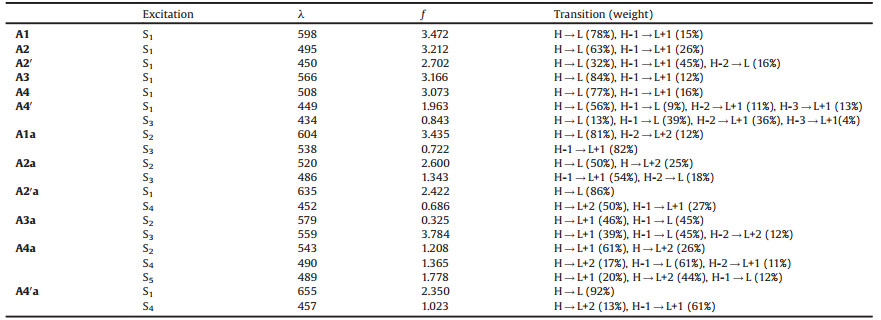
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

|
Download:
|
| Fig. 4. UV–vis absorption spectra and oscillator strengths of the excited states for the A-π-A acceptors. | |
{kind=link}
Because of additional "anti-aromatic" frontier molecular levels, more excitations can contribute to the absorption spectra of the "anti-aromatic" acceptors. For A1a, A2a, A3a, and A4a, the optically allowed transitions have similar energy gaps between the occupied and unoccupied orbitals (Fig. 3), so the corresponding excitation energies show small variation and are located near the S1 excitation of the "aromatic" acceptors. Hence the profiles of the absorption spectra for these "anti-aromatic" acceptors are similar to the related "aromatic" acceptors. However, owing to the additional contributions of the excitations from the "antiaromatic" levels, the absorption intensities are enhanced. Interestingly, for A2´a and A4´a, the energy gap is much smaller for the "anti-aromatic" HOMO→LUMO transition with respect to the other optically allowed transitions, arising from strong electronic coupling between the fused-ring units and the rest moieties for the "anti-aromatic" frontier orbitals. Consequently, besides the absorption at the wavelengths similar to A2´ and A4´, an extra absorption appears at much longer wavelengths; thus A2´a and A4a are hopeful to be a panchromatic sunlight absorber.
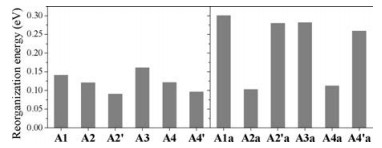
For organic solar cells, high charge carrier mobility will facilitate charge separation to improve short-circuit current, fill factor, and hence power conversion efficiency. Reorganization energy is one of the key parameters to determine charge transport performance in organic semiconductors; small reorganization energy is beneficial for achieving high mobility. Here, we are interested in the reorganization energies forelectron transport in theA-π-Aacceptors. The calculated results are shown in Fig. 5. The reorganization energies of A1 (IEIC) and A3 (ITIC) are ca. 0.14 eV and 0.16eV; these values are similar to those of fullerenes and twice smaller than those of perylenedii-mides [67]. When the fused thiophene on s-indacene is replaced by benzene, the reorganization energies are decreased for A2/A2´ and A4/A4´. Moreover, the decrease depends on the linking positions or fusion orientations of thiophene onto indacenodibenzene, and the A2´ and A4´ acceptors exhibit the smallest reorganization energies of < 0.1 eV. As seen in Fig. S8 (Supporting information), the reorganization energy decrease can be attributed to reduced variation of the bond lengths in the whole backbone upon charge.

|
Download:
|
| Fig. 5. Reorganization energies of electron transport for the A-p-A acceptors. | |
{kind=link}
Compared with A1 and A3, the reorganization energies for the "anti-aromatic" counterparts (A1a and A3a) are about twice increased, reaching ca. 0.3 eV. This can be due to big changes of the bond lengths in the anti-aromatic s-indacene core (Fig. S9 in Supporting information) since the electron accepting LUMO/ degenerate LUMO + 1 is completely localized on the s-indacene core for A1a/A3a (Fig. 2 and Fig. S6). Relatively, the (undegenerate) LUMO is more extended for A2´a and A4´a, so the reorganization energies are slightly decreased. In the case of A2a and A4a, the LUMO or degenerate LUMO +1 is delocalized over the electron-withdrawing INCN groups, leading to similar reorganization energies as the "aromatic" acceptors A2 and A4.
To summarize, we have theoretically studied the impact of π-bridge structures on optoelectronic properties of the A-π-A electron acceptors. In the case of the "aromatic" acceptors, incorporation of benzene instead of thiophene in the fused π-units will result in much deeper HOMO (dominated by the π-bridges) and slightly higher unoccupied frontier levels (dominated by the two terminal electron-withdrawing groups); thus the long-wavelength absorption, corresponding to the HOMO→LUMO transition, is significantly blue-shifted. At the same time, the reorganization energy for electron transport is decreased, which would be beneficial for improving the electron mobility. The calculations show that these optoelectronic properties are also very dependent on isomerization of the π-bridges, such as modification of linking or fusion modes of thiophene moieties. We underline that, anti-aromatization of the π-bridges will lead to additional and unique frontier levels for the "anti-aromatic" acceptors compared with the "aromatic" acceptors (Fig. 3). The multiplication of unoccupied frontier levels would be beneficial for achieving ultrafast "hot" charge separation [68, 69]. Moreover, the optical excitation energy for the transition between these additional "anti-aromatic" levels is in the proximity of or much smaller than that for the transition between the "aromatic" frontier levels for the different isomers. The optical absorption of the A-π-A acceptors is hence enhanced or broadened by anti-aromatization of the π-bridges. Our theoretical results point out the importance of π-bridge engineering, especially isomerization and anti-aromatization on tuning the electronic, optical, and electron transport properties of A-π-A electron acceptors. This would be very useful for the development of new electron acceptors for organic solar cells.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (No. 51773208), the Ministry of Science and Technology of China (No. 2014CB643506), and the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB12020200).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.05.029.
| [1] |
A.C. Arias, J.D. MacKenzie, I. McCulloch, J. Rivnay, A. Salleo, Chem. Rev. 110 (2010) 3-24. DOI:10.1021/cr900150b |
| [2] |
R. Søndergaard, M. Hösel, D. Angmo, T.T. Larsen-Olsen, F.C. Krebs, Mater. Today 15 (2012) 36-49. DOI:10.1016/S1369-7021(12)70019-6 |
| [3] |
M. Kaltenbrunner, M.S. White, E.D. Głowacki, et al., Nat. Commun. 3 (2012) 770. DOI:10.1038/ncomms1772 |
| [4] |
K. Liu, T.T. Larsen-Olsen, Y. Lin, et al., J. Mater. Chem. A 4 (2016) 1044-1051. DOI:10.1039/C5TA07357J |
| [5] |
X. Gu, Y. Zhou, K. Gu, et al., Adv. Energy Mater. 7 (2017) 1602742. DOI:10.1002/aenm.201602742 |
| [6] |
Y. W. Su, S.C. Lan, K.H. Wei, Mater. Today 15 (2012) 554-562. DOI:10.1016/S1369-7021(13)70013-0 |
| [7] |
C.W. Tang, Appl. Phys. Lett. 48 (1986) 183-185. DOI:10.1063/1.96937 |
| [8] |
G. Yu, J. Gao, J.C. Hummelen, F. Wudl, A.J. Heeger, Science 270 (1995) 1789-1791. DOI:10.1126/science.270.5243.1789 |
| [9] |
P.W.M. Blom, V.D. Mihailetchi, L.J.A. Koster, D.E. Markov, Adv. Mater. 19 (2007) 1551-1566. DOI:10.1002/(ISSN)1521-4095 |
| [10] |
B. Kippelen, J. L. Bredas, Energy Environ. Sci. 2 (2009) 251-261. DOI:10.1039/b812502n |
| [11] |
T.M. Clarke, J.R. Durrant, Chem. Rev. 110 (2010) 6736-6767. DOI:10.1021/cr900271s |
| [12] |
A.J. Heeger, Adv. Mater. 26 (2014) 10-28. DOI:10.1002/adma.201304373 |
| [13] |
B. Kan, M. Li, Q. Zhang, et al., J. Am. Chem. Soc. 137 (2015) 3886-3893. DOI:10.1021/jacs.5b00305 |
| [14] |
Z. He, B. Xiao, F. Liu, et al., Nat. Photon. 9 (2015) 174-179. DOI:10.1038/nphoton.2015.6 |
| [15] |
Q. Zhang, B. Kan, F. Liu, et al., Nat. Photon. 9 (2014) 35-41. |
| [16] |
Z. Li, K. Jiang, G. Yang, et al., Nat. Commun. 7 (2016) 13094. DOI:10.1038/ncomms13094 |
| [17] |
D. Deng, Y. Zhang, J. Zhang, et al., Nat. Commun. 7 (2016) 13740. DOI:10.1038/ncomms13740 |
| [18] |
J. Zhao, Y. Li, G. Yang, et al., Nat. Energy 1 (2016) 15027. DOI:10.1038/nenergy.2015.27 |
| [19] |
D. Meng, D. Sun, C. Zhong, et al., J. Am. Chem. Soc. 138 (2016) 375-380. DOI:10.1021/jacs.5b11149 |
| [20] |
D. Meng, H. Fu, C. Xiao, et al., J. Am. Chem. Soc. 138 (2016) 10184-10190. DOI:10.1021/jacs.6b04368 |
| [21] |
M. Li, K. Gao, X. Wan, et al., Nat. Photon. 11 (2016) 85. |
| [22] |
L. Yang, S. Zhang, C. He, et al., J. Am. Chem. Soc. 139 (2017) 1958-1966. DOI:10.1021/jacs.6b11612 |
| [23] |
H. Bin, Y. Yang, Z. G. Zhang, et al., J. Am. Chem. Soc. 139 (2017) 5085-5094. DOI:10.1021/jacs.6b12826 |
| [24] |
Z.G. Zhang, Y. Yang, J. Yao, et al., Angew. Chem. Int. Ed. 56 (2017) 13503-13507. DOI:10.1002/anie.201707678 |
| [25] |
B. Fan, L. Ying, P. Zhu, et al., Adv. Mater. 29 (2017) 1703906. DOI:10.1002/adma.201703906 |
| [26] |
B. Fan, L. Ying, Z. Wang, et al., Energy Environ. Sci. 10 (2017) 1243-1251. DOI:10.1039/C7EE00619E |
| [27] |
D. Baran, R.S. Ashraf, D.A. Hanifi, et al., Nat. Mater. 16 (2016) 363-369. |
| [28] |
Y. Duan, X. Xu, H. Yan, et al., Adv. Mater. 29 (2016) 1605115. |
| [29] |
W. Zhao, S. Li, H. Yao, et al., J. Am. Chem. Soc. 139 (2017) 7148-7151. DOI:10.1021/jacs.7b02677 |
| [30] |
M.C. Scharber, D. Mühlbacher, M. Koppe, et al., Adv. Mater. 18 (2006) 789-794. DOI:10.1002/(ISSN)1521-4095 |
| [31] |
B.C. Thompson, J.M.J. Fréchet, Angew. Chem. Int. Ed. 47 (2008) 58-77. DOI:10.1002/(ISSN)1521-3773 |
| [32] |
J. Brabec Christoph, S. Gowrisanker, J.M. Halls Jonathan, et al., Adv. Mater. 22 (2010) 3839-3856. DOI:10.1002/adma.200903697 |
| [33] |
Y. He, H.Y. Chen, J. Hou, Y. Li, J. Am. Chem. Soc. 132 (2010) 1377-1382. DOI:10.1021/ja908602j |
| [34] |
Y. He, Y. Li, Phys. Chem. Chem. Phys. 13 (2011) 1970-1983. DOI:10.1039/C0CP01178A |
| [35] |
Y. Lin, X. Zhan, Mater. Horiz. 1 (2014) 470-488. DOI:10.1039/C4MH00042K |
| [36] |
M.C. Scharber, Adv. Mater. 28 (2016) 1994-2001. DOI:10.1002/adma.201504914 |
| [37] |
C.B. Nielsen, S. Holliday, H.Y. Chen, S.J. Cryer, I. McCulloch, Acc. Chem. Res. 48 (2015) 2803-2812. DOI:10.1021/acs.accounts.5b00199 |
| [38] |
N. Liang, W. Jiang, J. Hou, Z. Wang, Mater. Chem. Front. 1 (2017) 1291-1303. DOI:10.1039/C6QM00247A |
| [39] |
J. Hou, O. Inganäs, R.H. Friend, F. Gao, Nat. Mater. 17 (2018) 119-128. DOI:10.1038/nmat5063 |
| [40] |
Y. Lin, J. Wang, Z. G. Zhang, et al., Adv. Mater. 27 (2015) 1170-1174. DOI:10.1002/adma.201404317 |
| [41] |
Y. Lin, Z.G. Zhang, H. Bai, et al., Energy Environ. Sci. 8 (2015) 610-616. DOI:10.1039/C4EE03424D |
| [42] |
H. Yao, Y. Cui, R. Yu, et al., Angew. Chem. Int. Ed. 56 (2017) 3045-3049. DOI:10.1002/anie.201610944 |
| [43] |
R. Yu, S. Zhang, H. Yao, et al., Adv. Mater 29 (2017) 1700437. DOI:10.1002/adma.v29.26 |
| [44] |
Y. Cui, H. Yao, B. Gao, et al., J. Am. Chem. Soc. 139 (2017) 7302-7309. DOI:10.1021/jacs.7b01493 |
| [45] |
X. Xu, T. Yu, Z. Bi, et al., Adv. Mater. 30 (2017) 1703973. |
| [46] |
R. Breslow, B. Jaun, R.Q. Kluttz, C.Z. Xia, Tetrahedron 38 (1982) 863-867. DOI:10.1016/0040-4020(82)80167-1 |
| [47] |
A. Minsky, A.Y. Meyer, M. Rabinovitz, Tetrahedron 41 (1985) 785-791. DOI:10.1016/S0040-4020(01)96458-0 |
| [48] |
L. Qiu, X. Zhuang, N. Zhao, et al., Chem. Commun. 50 (2014) 3324-3327. DOI:10.1039/C3CC49418G |
| [49] |
G. Dai, J. Chang, X. Shi, et al., Chem.-Eur. J. 21 (2014) 2019-2028. |
| [50] |
Z. Zeng, X. Shi, C. Chi, et al., Chem. Soc. Rev. 44 (2015) 6578-6596. DOI:10.1039/C5CS00051C |
| [51] |
J. Zheng, X. Zhuang, L. Qiu, et al., J. Phys. Chem. A 119 (2015) 3762-3769. |
| [52] |
Chaolumen, M. Murata, A. Wakamiya, Y. Murata, Org. Lett. 19 (2017) 826-829. DOI:10.1021/acs.orglett.6b03819 |
| [53] |
A. Mailman, A.A. Leitch, W. Yong, et al., J. Am. Chem. Soc. 139 (2017) 2180-2183. DOI:10.1021/jacs.6b12814 |
| [54] |
D.T. Chase, A.G. Fix, B.D. Rose, et al., Angew. Chem. Int. Ed. 50 (2011) 11103-11106. DOI:10.1002/anie.v50.47 |
| [55] |
J.L. Marshall, K. Uchida, C.K. Frederickson, et al., Chem. Sci. 7 (2016) 5547-5558. DOI:10.1039/C6SC00950F |
| [56] |
R. Grollman, N. Quist, A. Robertson, et al., J. Phys. Chem. C 121 (2017) 12483-12494. DOI:10.1021/acs.jpcc.7b03729 |
| [57] |
A.G. Fix, P.E. Deal, C.L. Vonnegut, et al., Org. Lett. 15 (2013) 1362-1365. DOI:10.1021/ol400318z |
| [58] |
H. Miyoshi, S. Nobusue, A. Shimizu, et al., Chem. Sci. 5 (2014) 163-168. DOI:10.1039/C3SC52622D |
| [59] |
J. Cao, G. London, O. Dumele, et al., J. Am. Chem. Soc. 137 (2015) 7178-7188. DOI:10.1021/jacs.5b03074 |
| [60] |
J.J. Dressler, Z. Zhou, J.L. Marshall, et al., Angew. Chem. Int. Ed. 56 (2017) 15363-15367. DOI:10.1002/anie.201709282 |
| [61] |
D.T. Chase, A.G. Fix, S.J. Kang, et al., J. Am. Chem. Soc. 134 (2012) 10349-10352. DOI:10.1021/ja303402p |
| [62] |
J. I. Nishida, S. Tsukaguchi, Y. Yamashita, Chem.-Eur. J. 18 (2012) 8964-8970. DOI:10.1002/chem.v18.29 |
| [63] |
L. Ren, C. Liu, Z. Wang, X. Zhu, J. Mater. Chem. C 4 (2016) 5202-5206. DOI:10.1039/C6TC01808D |
| [64] |
I. Martinez, E. Schott, I. Chávez, J.M. Manríquez, X. Zarate, Chem. Phys. Lett. 659 (2016) 31-35. DOI:10.1016/j.cplett.2016.06.079 |
| [65] |
I. Martinez, X. Zarate, E. Schott, et al., Chem. Phys. Lett. 636 (2015) 31-34. DOI:10.1016/j.cplett.2015.06.085 |
| [66] |
H.U. Kim, J. H. Kim, H. Suh, et al., Chem. Commun. 49 (2013) 10950-10952. DOI:10.1039/c3cc46557h |
| [67] |
Y. Yi, V. Coropceanu, J. L. Bredas, J. Mater. Chem. 21 (2011) 1479-1486. DOI:10.1039/c0jm02467h |
| [68] |
X. Shen, G. Han, D. Fan, Y. Xie, Y. Yi, J. Phys. Chem. C 119 (2015) 11320-11326. |
| [69] |
A. Kuzmich, D. Padula, H. Ma, A. Troisi, Energy Environ. Sci. 10 (2017) 395-401. DOI:10.1039/C6EE03654F |