 2019, Vol. 30
2019, Vol. 30 


b ICQD/Hefei National Laboratory for Physical Sciences at Microscale, and Key Laboratory of Strongly-Coupled Quantum Matter Physics, Chinese Academy of Sciences and Department of Physics, University of Science and Technology of China, Hefei 230026, China;
c Key Laboratory of Optoelectronic Devices and Systems of Ministry of Education and Guangdong Province, Shenzhen University, Shenzhen 518060, China;
d Shenzhen Research Institute of Hunan University, Shenzhen 518057, China
Methanol has been used as a clean energy resource to tackle global energy shortage and environmental pollution problems [1-4]. However, the methanol gas is highly poisonous, and inhaling the high concentrations of methanol gas or absorpting of methanol through the skin is often extremely harmful to human beings [5]. For these reasons, it is especially necessary to develop a reliable and selective methanol gas sensor. In the past few years, there has been performed extensive work on methanol adsorption and decomposition, or methanol gas sensing based on different materials with various surface, such as ZnO(1010) surface [1], PtRuPt(111) [2], Ru(0001) surfaces [3], MoS2 surface [4], Ni(Pd)-decorated grapheme [6], graphene supported Pt13 nanoclusters [7], TbOx(111) surfaces [8], Pt3Ni(111) surface [9] and Al-doped ZnO thin films [10].
Most recently, phosphorene (single layer black phosphorus) has been successfully fabricated by mechanical cleavage from black phosphorus [11]. Similar to graphene and MoS2, phosphorene is a single layered two-dimensional (2D) nanomaterial [12]. Theoretical calculation revealed that phosphorene is an intrinsic p-type semiconductor material with the direct band gap of about 0.73 eV [13, 14]. Besides, phosphorene possesses a large free-carrier mobility (up to 1000cm2V-1 s-1), good mechanical flexibility, linear dichroism, optical responses and high on/off ratios (up to 104 mobility) [15-19]. And more significantly, phosphorene has an anisotropic electric conductance and is sensitive to environmental gas molecules [16-19], thus has many promising applications in nanoelectronic devices. Therefore, many experiments and theoretical calculations have been performed to study the adsorption and dissociation of small gas molecules over phosphorene surface. Zhang and Jing reported the first-principle study of the adsorption of tetracyano-p-quinodimethane (TCNQ) and tetrathiafulvalene (TTF) molecule on phosphorene [12, 20]. Cai researched the physisorbed adsorption of CO, H2, H2O, NH3, NO, NO2, and O2 on phosphorene [21]. Kulish surveyed the metal adatoms on single-layer phosphorene based on density functional theory [22]. Yu, Arabieha, Lalitha and Kuanga further investigated the gases adsorption of heteroatom-doped phosphorene [16-18, 23]. In 2015, Carmen researched methanol gas sensing based on layered black phosphorus using electrochemical impedance spectroscopy as the detection method [5]. The results indicated that the sensing for methanol detection based on black phosphorus is high-efficiency and cost-effective. However, there are no studies about the adsorption properties of methanol gas molecule over phosphorene surface by means of the density functional theory calculation.
Herein, we firstly presented the study of the adsorption properties of methanol gas molecule on pristine and X-doped phosphorene (X = B, C, N and O) based on first-principle calculations. In the adsorption models, the CH3OH gas molecule was placed onthe top of different phosphorenes, O—H bond of CH3OH was parallel to phosphorene surfaces, and the whole adsorption systems were fully optimized byusingVienna ab initio simulation package (VASP). Our results show that the adsorption of CH3OH gas molecule on N-doped and O-doped phosphorene is more efficient compared to the pristine phosphorene. However, B-doped and C-doped phosphor-ene are almost not beneficial. Our present study provides a valuable reference for methanol gas adsorption and sensor based on phosphorene-based material.
Our calculations are performed based on the density functional theory (DFT) and electron-ion interactions are investigated with the use of the projector augmented wave (PAW) method, which implemented in the VASP [13, 14]. The exchange corrective interaction is described bythe generalized gradient approximation (GGA) with the parametrization of Perde-Burke-Ernzerhof (PBE) and we perform non spin-polarized calculations for all of the structural optimizations [24]. The van der Waals density function (vdW-DF) of Becke88 optimization (optB88) is employed to improve the description of the PBE function [13, 14]. A planewave basis set with an energy cutoff of 450 eV is employed for the planewave expansion of the wave function. The atomic positions are fully relaxed until satisfying an energy convergence of 1 × 10-4eV and force convergence of 0.05eV/Å.
We adapt a 3 × 3 phosphorene supercell along the x and y directions of the phosphorene layer. The lattice constant of the primitive unit cell is a = 3.32Å, b = 4.32Å, and 36 atoms are contained. In order to safely avoid the interaction between the periodically repeated structures, a vacuum thickness of 20Å between the layers is used. Thus, the methanol-phosphorene adsorption systems are composed of a 10.04Å × 13.29 Å × 22.16 Å supercell. The first Brillouin zone is sampled using a Monkhorst-Pack (MP) grid corresponding to 5 × 1 × 5 k-point mesh for both geometry optimization and electronic properties calculations. In order to estimate the charge distribution and corresponding charge transfer (ΔQ) between the CH3OH gas molecule and phosphorene, Bader charge analysis method is adopt [17, 20, 22, 25].
In order to characterize the stability of the CH3OH gas molecule adsorption on phosphorene, the adsorption energy is determined as:

|
(1) |
where Ephosphorene+CH3OH, Ephosphorene and ECH3OH stand for the total energy of the molecular adsorption on phosphorene, the isolated phosphorene layer and the single CH3OH molecule, respectively. According to this definition, a more negative Ea indicates a more favorable interaction.
To better understand the nature of the adsorption mechanism, the isosurface of the charge density difference (CDD) Δρ of the methanol-phosphorene adsorption systems is defined as:

|
(2) |
where ρphosphorene+CH3OH, ρphosphorene and ρCH3OH stand for the charge distribution of the molecular adsorption on phosphorene, the isolated phosphorene layer and the single CH3OH molecule respectively. According to this definition, a positive ΔQ indicates the transfer of electrons from CH3OH molecule to phosphorene. charge distribution of the molecular adsorption on phosphorene, the isolated phosphorene layer and the single CH3OH molecule respectively. According to this definition, a positive DQ indicates the transfer of electrons from CH3OH molecule to phosphorene.
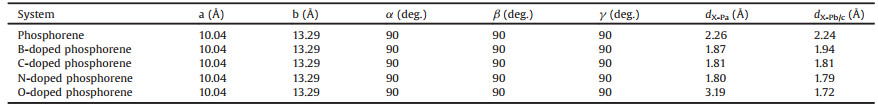
The optimized geometry of single layer phosphorene from both the top and side views is shown in Fig. 1. The relaxed lattice constant is a = 3.35 Å and b = 4.43 Å along the armchair and zigzag directions. The calculated P—P bonds length in and out of the plane are 2.24 Å and 2.26 Å, and the corresponding bond angles are 96.5° and 102.3°, respectively. These are in good agreement with the previous theoretical studies [13, 14].

|
Download:
|
| Fig. 1. The top and side views of the optimized geometry of single layer phosphorene and three adsorption sites: the hollow site (H), the bridge site (B), and the top site (T). | |
The electronic properties of the single layer phosphorene are investigated through the density of states (DOS) and the band structure, which are shown in Fig. S1 (Supporting information). The energy gap of the single layer phosphorene is about 0.73 eV, and the valance band minima (VBM) and conduction band maxima (CBM) are both located at the high symmetry point in the reciprocal space. These reveal that phosphorene is a direct band-gap semiconductor, which is in agreement with previous researches [13, 14, 20].
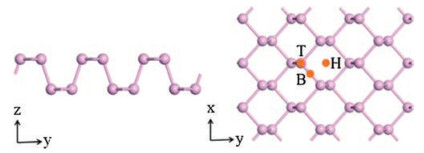
The pristine phosphorene has sp3 hybridization, where P bonds with three adjacent P with a lone pair of electrons, resulting in a puckered structure [14-16]. Fig. 2 shows the schematic illustration of top and side views of the optimized X-doped phosphorene structures (X = B, C, N and O), and Table 1 shows the calculated results of lattice constants, bond lengths and distances between two atoms in pristine and X-doped phosphorene, respectively. Here, we define P atom on a, b and c sites as Pa, Pb and Pc respectively. In pristine phosphorene, dx-Pa and dx-Pb/c is 2.26 Å and 2.24Å. The X-Pa distances in B, C, N and O-doped phosphorene systems are found to be 1.87, 1.81, 1.80 and 3.19A, and X-Pb/c distances are 1.94, 1.81, 1.79 and 1.72Å respectively, which agree with the previous calculations [18, 26, 27].

|
Download:
|
| Fig. 2. The schematic illustration of top and side views of optimized X-dopedphosphorene structures (X = B, C, N and O). | |
|
|
Table 1 The calculated results of lattice constants, bond lengths and distances between two atoms in pristine and X-doped phosphorene, respectively. |
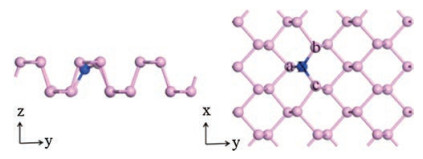
We then discuss the adsorption of CH3OH gas molecule on single layer phosphorene and heteroatom-doped phosphorene. Three adsorption sites are considered as Fig. 1 shows: the hollow site (H), the bridge site (B), and the top site (T) [22, 28]. In the adsorption models, the CH3OH gas molecule is placed on the top of different phosphorenes, O—H bond of CH3OH is parallel to phosphorene surfaces, and the whole systems are fully optimized by using VASP. Fig. 3 shows the most stable methanol-phosphor-ene adsorption configurations. For abetter understanding of the adsorption of CH3OH gas molecule on these different phosphorene surfaces, the adsorption sites, adsorption distances (d) between CH3OH gas molecule and the phosphorene surface and their corresponding adsorption energies (Ea) (Eq. (1)) are calculated and listed in Table 2. The d refers to the distance between oxygen atom of CH3OH molecule and the surface of phosphorene or heteroatom-doped phosphorene. It can be observed that the CH3OH molecule's orientations are almost unchanged and the adsorption distances vary from 2.1 Å to 2.83 Å. For the pristine phosphorene, the adsorption energy is -0.24eV and the adsorption distance is 2.74Å, which indicates the adsorption behavior is allowed in thermodynamics and the gas molecule is physically absorbed on the phosphorene surface. This is agree with the previous experimental results [6, 12, 16, 21]. Moreover, the adsorption energies between CH3OH with B-doped, C-doped, N-doped and O-doped phosphorene are -0.26, -0.25, -0.29 and -0.23 eV, and the corresponding distances are 2.78, 2.83, 2.51 and 2.10 Å, respectively. The adsorption energies and corresponding distances between CH3OH with B-doped and C-doped samples are similar to the pristine phosphorene. However, N-doped phosphorene possess the significantly shorter adsorption distance and largest adsorption energy. O-doped phosphorene has the shortest adsorption distance although the adsorption energy is close to the pristine phosphorene. These indicate N-doped and O-doped phosphorene are more effective for the adsorption ofCH3OH gas molecule due to the shorter adsorption distance and larger adsorption energy, while B-doped and C-doped phosphorene are almost not beneficial compared to the pristine phosphorene.

|
Download:
|
| Fig. 3. The top and side views of the most stable methanol adsorption configurations on (a) pristine, (b) B-doped, (c) C-doped, (d) N-doped and (e) O-doped phosphorene. | |
|
|
Table 2 The adsorption sites, adsorption distances and adsorption energies for methanol adsorption on the pristine phosphorene and heteroatom-doped phosphorene after relaxation. |

{kind=link}
{kind=link}
{kind=link}
In order to estimate the Δρ (Eq. (2)) and corresponding charge transfer (ΔQ) between CH3OH gas molecule and phosphorene, Bader charge analysis is implemented as shown in Fig. 4.

|
Download:
|
| Fig. 4. Charge density difference (CDD) for the adsorption of CH3OH gas molecule on (a) pristine, (b) B-doped, (c) C-doped, (d) N-doped and (e) O-doped phosphorene. The isosurface value is 0.001 e/Å3. The yellow regions show the charge accumulation, whereas the blue regions represent the charge depletion. | |
{kind=link}
For the O-doped phosphorene, the amount of electrons transferred from CH3OH gas molecule to O-doped phosphorene is the largest (up to 0.04 e), which leads to the shortest adsorption distance (the adsorption energy is not the largest). Also, the N-doped phosphorene has larger charge transfer (near 0.03 e) with corresponding shorter adsorption distance and larger adsorption energy when compared to the pristine phosphorene.
However, the amount of the transferred electrons for B-doped and C-doped phosphorene are obviously smaller (about 0.02 e), which results in the longer distances and similar adsorption energies compared to the pristine phosphorene. Furthermore, it could be observed from the charge density difference that the charges are remarkably redistributed after adsorbing CH3OH gas molecule and the charge is transfered from CH3OH to N-doped or O-doped phosphorene. This gives a further explanation that N-doped and O-doped phosphorene are more effective for the adsorption of CH3OH gas molecule.
In conclusion, we have firstly performed a theoretical research on the adsorption properties between methanol gas molecule and pristine or heteroatom-doped phosphorene by using VASP. The calculation results show that N-doped and O-doped phosphorene have larger charge transfer values with corresponding shorter adsorption distances and larger adsorption energies for the adsorption of methanol compared to the pristine phosphorene, while B-doped and C-doped phosphorene are not desired. Our results suggest that N-doped and O-doped phosphorene are ideal candidates when used for CH3OH gas sensing.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21701043, 21573066, 51402100), the Provincial Natural Science Foundation of Hunan (Nos. 2016JJ1006, 2016TP1009), the Key Laboratory of Optoelectronic Devices and Systems of Ministry of Education and Guangdong Province and Shenzhen Science and Technology Program (No. JCYJ20170306141659388).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/jxclet.2018.01.041.
| [1] |
C.T. Vo, L.K. Huynh, J.Y. Hung, J.C. Jiang, Appl. Surf. Sci. 280 (2013) 219-224. DOI:10.1016/j.apsusc.2013.04.135 |
| [2] |
Q. Ding, W. Xu, P. Sang, et al., Appl. Surf. Sci. 369 (2016) 257-266. DOI:10.1016/j.apsusc.2015.11.104 |
| [3] |
J. Liu, C. Lü, C. Jin, Y. Guo, G. Wang, Chem. Res. Chin. Univ. 32 (2016) 234-241. DOI:10.1007/s40242-016-5416-z |
| [4] |
Y.Y. Chen, M. Dong, Z. Qin, et al., J. Mol. Catal. A:Chem. 338 (2011) 44-50. |
| [5] |
C.C. Mayorga-Martinez, Z. Sofer, M. Pumera, Angew. Chem. Int. Ed. 54 (2015) 14317-14320. DOI:10.1002/anie.201505015 |
| [6] |
A.A. Peyghan, M. Moradi, J. Iran. Chem. Soc. 12 (2014) 1-6. |
| [7] |
R.J. Gasper, A. Ramasubramaniam, J. Phys. Chem. C 120 (2016) 17408-17417. DOI:10.1021/acs.jpcc.6b04557 |
| [8] |
A. Schaefer, W.C. Cartas, R. Rai, et al., J. Phys. Chem. C 120 (2016) 28617-28629. DOI:10.1021/acs.jpcc.6b09908 |
| [9] |
P. Du, P. Wu, C. Cai, J. Phys. Chem. C 121 (2017) 9348-9360. |
| [10] |
P.P. Sahay, R.K. Nath, Sensor. Actuat. B-Chem. 134 (2008) 654-659. |
| [11] |
E.S. Reich, Nature 506 (2014) 7486. |
| [12] |
R. Zhang, B. Li, J. Yang, J. Phys. Chem. C 119 (2015) 2871-2878. DOI:10.1021/jp5116564 |
| [13] |
J. Qiao, X. Kong, Z.X. Hu, F. Yang, W. Ji, Nat. Commun. 5 (2014) 4475. DOI:10.1038/ncomms5475 |
| [14] |
M.Z. Rahman, W.K. Chi, K. Davey, S.Z. Qiao, Energy Environ. Sci. 9 (2016) 1513-1514. DOI:10.1039/C6EE90016J |
| [15] |
A. Srivastava, M.S. Khan, S.K. Gupta, R. Pandey, Appl. Surf. Sci. 356 (2015) 881-887. DOI:10.1016/j.apsusc.2015.08.109 |
| [16] |
M. Lalitha, Y. Nataraj, S. Lakshmipathi, Appl. Surf. Sci. 377 (2016) 311-323. DOI:10.1016/j.apsusc.2016.03.119 |
| [17] |
A. Kuang, M. Kuang, H. Yuan, et al., Appl. Surf. Sci. 410 (2017) 505-512. DOI:10.1016/j.apsusc.2017.03.135 |
| [18] |
M. Arabieh, Y.T. Azar, Appl. Surf. Sci. 396 (2017) 1411-1419. DOI:10.1016/j.apsusc.2016.11.175 |
| [19] |
Q. Yang, R.S. Meng, J.K. Jiang, et al., IEEE Electron Device Lett. 37 (2016) 660-662. DOI:10.1109/LED.2016.2543243 |
| [20] |
Y. Jing, Q. Tang, P. He, Z. Zhou, P. Shen, Nanotechnology 26 (2015) 095201. DOI:10.1088/0957-4484/26/9/095201 |
| [21] |
Y. Cai, Q. Ke, G. Zhang, Y.W. Zhang, J. Phys. Chem. C 119 (2015) 3102-3110. DOI:10.1021/jp510863p |
| [22] |
V.V. Kulish, O.I. Malyi, C. Persson, P. Wu, Phys. Chem. Chem. Phys. 17 (2015) 992-1000. DOI:10.1039/C4CP03890H |
| [23] |
Z. Yu, N. Wan, S. Lei, H. Yu, J. Appl. Phys. 120 (2016) 024305. DOI:10.1063/1.4958695 |
| [24] |
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865-3868. DOI:10.1103/PhysRevLett.77.3865 |
| [25] |
N. Suvansinpan, F. Hussain, G. Zhang, et al., Nanotechnology 27 (2016) 065708. DOI:10.1088/0957-4484/27/6/065708 |
| [26] |
L. Yang, W. Mi, X. Wang, J. Alloys Compd. 662 (2016) 528-533. DOI:10.1016/j.jallcom.2015.12.095 |
| [27] |
W. Yu, Z. Zhu, C.Y. Niu, et al., Phys. Chem. Chem. Phys. 17 (2015) 16351-16358. DOI:10.1039/C5CP01732G |
| [28] |
T. Hu, J. Hong, J. Phys. Chem. C 119 (2015) 8199-8207. DOI:10.1021/acs.jpcc.5b01300 |