 2019, Vol. 30
2019, Vol. 30 


b Department of Polymer Science and Engineering, School of Chemistry and Chemical Engineering, Hefei University of Technology and Anhui Key Laboratory of Advanced Catalytic Materials and Reaction Engineering, Hefei 230009, China
Most of the biological macromolecules like polysaccharides [1], protein [2] and DNA [3] are optically active and some ofthem present characteristic functionalities such as catalysing metabolic reactions, molecular recognition, structure-related and stores biological information. Inspired by biological helices and their elaborate functions, chemists have been challenged to develop artificial helical oligomers and polymers for potential applications in materials science, such as ferroelectric liquid crystals, nonlinear optical materials, sensing specific molecules, the separation ofenantiomers and asymmetric catalysis [4]. Chiral compounds show specific structure-activity relationships, meanwhile racemic mixtures and individual stereoisomers can differ significantly in living systems [5]. Therefore, enantiomer separation from racemic mixtures is an important process that can now be accomplished with chiral columns [6, 7], membrane filtration [8], asymmetric catalysis [9], enantiomer-selective crystallization [10, 11] and enantiomer-selective polymerization [12, 13]. Enantiomer-selective polymerization means that one enantiomer is preferentially polymerized to obtain opticallyactive polymerand unreacted monomer. It is a resolutionof kinetic and stereospecific [14]. Enantiomer-selectivity can be achieved byopticallyactive catalysts or initiators. Eventhe preferred helical structure of the polymer chain can also contribute to the enantiomer-selectivity during the polymerization [15]. Allene is a special kind of monomer with interesting characteristic which allows them to obtain polymers through the selective polymerization of either part (1, 2- or 2, 3-) of cumulative double bond [16, 17]. Although investigations about polyallene have been carried out in recent decades, the study ofoptical activity is still limited [18, 19].The allylnikel(Ⅱ) catalysts is a stable and highly chemoselective complex, which has been used for living polymerization of allenes containing various functional groups [20-22]. Due to the interaction between the complex and monomers at the active end of living chain, the coordination polymerization has a good performance on the enantiomer-selectivity [23]. One of the strategies for enantiomer- selective polymerization is coordination polymerization with a chiral ligand complex catalyst [24, 25]. We believe the present study provides a clue for choosing chiral ligand to explore the possibility of enantiomer-selectivity for chiral allene monomer. In order to adjust the environment of the coordination polymerization, the PPh3 in the allylnickel(Ⅱ) complex catalyst was replaced by chiral phosphine.
In this contribution, the living and enantiomer-selective polymerization of allene derivatives initiated by 2 was reported. First, 2 was prepared according to a previously reported method by our group with slightly modification [26]. The living nature of the polymerization of L-1 with 2 was investigated. Polymerization in different initial feed ratios of L-1 to 2 were then performed (Scheme 1). The Mns and Mw/Mn values of obtained polymers were analyzed by size exclusion chromatography (SEC) with equivalent to polystyrene standards. As SEC traces shown in Fig. 1a, all the obtained polymers exhibited a single-modal elution peak. The Mn increased linearly with the conversion of L-1 and all the obtained polymers showed narrow molecular weight distributions with Mw/Mn < 1.24 (Fig. 1b). Thus, it was confirmed that the polymerizations of L-1 using 2 as an initiator proceeded in a living/controlled manner.

|
Download:
|
| Scheme 1. The preparation of poly-L-1100, poly(L-150-b-D-150) and poly(L-150-b-rac-150). | |

|
Download:
|
| Fig. 1. (a) SEC chromatograms of poly-L-1m prepared in CH2Cl2 with different initial mfeed ratios of L-1 to 2 at 298 K. (b) Mn and Mw/Mn values as functions of conversion of L-1 to prepare poly-L-150. | |
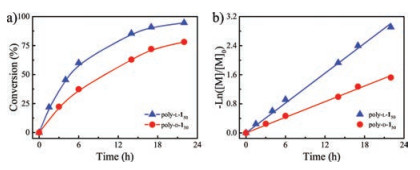
To get a deeper insight into this new polymerization system, kinetic studies of L-1 and D-1 with 2 were carried out. The polymerization was conducted in the presence of an internal dimethyl terephthalate (DMT) as standard. The reactions were followed by 1H NMR measurements of the aliquots taken out from the polymerization system at appropriate time intervals to follow the changes in the relative relationship of L-1 or D-1 with respect to DMT to calculate the monomer conversions. The conversion of L-1 and D-1 in polymerization was shown in Fig. 2a, which clearly revealed that the polymerization rate of L-1 was faster than that of the enantiomeric antipode D-1.

|
Download:
|
| Fig. 2. (a) Plots of the conversionwith the polymerization time of monomer L-1 and D-1 initiated by 2 in CH2O2 at 298 K. (b) First-order kinetic plots for the polymerization of L-1 and D-1 initiated by 2 in CH2O2 at 298 K. | |
As summarized in Fig. 2b, the linear correlation between -ln ([M]/[M]0) and the polymerization time revealed that the two reactions obeyed first-order rate law. The consumption rate of L-1 (kL-1) was faster than that of D-1 (kD-1). The kL-1 and kD-1 were calculated as 3.80 × 10-5 and 1.99 × 10-5s-1 respectively. It was worthwhile to note that the reaction rates were much slower than that of using PPh3 complex catalyst [26]. Kinetic studies showed that L-1 was preferentially polymerized over the antipode D-1 by a factor of 1.9. These results indicated that the chiral Ni complex affected the addition of chiral monomer to the growing end. It can be attributed to the coordination structure around the metal center with the chiral ligand, which provided a rigid and asymmetric environment making the reaction affected. The addition of steric congestion at the metal center slowed the polymer formation. The difference in chiral compatibility between catalyst and two enantiomers made their reaction rate different. The obvious difference between kL-1 and kD-1 provided the possibility for the enantiomer-selectivity of allene bearing chiral amide pendants.
Then, using Ni(Ⅱ)-terminated poly-L-150 as macroinitiator, poly-L-1100, poly(L-150-b-D-150)and poly(L-150-b-rac-150)wereobtained via sequential addition of monomer L-1, D-1 and rac-1 respectively. As shown in Table 1, Ni(Ⅱ)-terminated poly-L-150 can initiate a living/controlled block copolymerization.
|
|
Table 1 Mn and Mw/Mn values of the copolymer sampling form the polymerization system of preparing poly(L-150-b-D-150) and poly(L-150-b-rac-150). |

{kind=link}
{kind=link}
{kind=link}
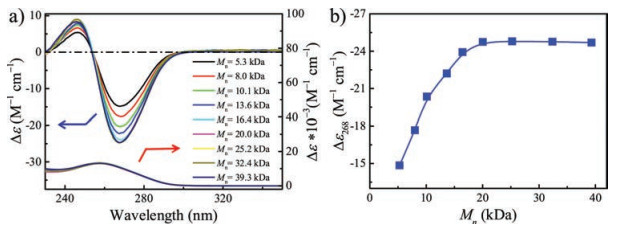
Next, the optical activity changes of polymers were studied. Amides are widely used in the design and preparation offunctional materials. Chirality and intermolecular hydrogen bonding are essential to the stable helical conformation with a preferred handedness. As shown in Fig. 3a, chiral amide-based polyallenes presented stable helix structure in THF.

|
Download:
|
| Fig. 3. (a) Molar circular dichroism and UV-vis spectra of poly-L-1m measured inTHF at 298 K. (b) The plot of molar circular dichroism of poly-L-1m measured inTHF at 298 K as a function of Mn (c = 0.03 g/L). | |
{kind=link}
The molar circular dichroism of the poly-L-1ms with different Mn was shown in Fig. 3b. When Mn was increased in the range of 5.3 kDa to 20.0 kDa, the corresponding molar circular dichroism gradually became larger and reached the maximum value of -24.7 L mol-1 cm-1. 20.0 kDa was a critical value of Mn. When the Mn was above the value, the molar circular dichroism no longer varied. This indicated that the helical structure with preferred handedness of poly-L-150 (Mn = 20.0kDa) gradually stabilized, and no longer changing as repeating unit elongated. That's why we choose poly-L-150 as a macroinitiator.
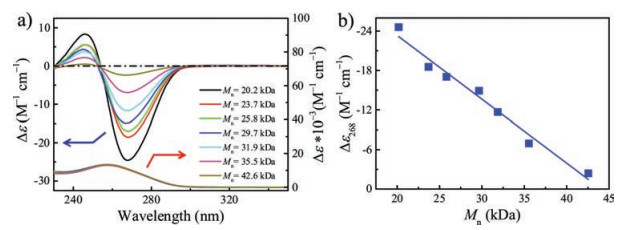
The samples taken from the copolymerization system of preparing poly(L-150-b-D-150) and poly(L-150-b-rac-150) were analyzed. For two copolymerization systems, the Mns of macroinitiator were both close to 20.0 kDa. They also had similar molar circular dichroism of -24.7 L mol-1 cm-1 and helical structure. As shown in Fig. 3b, it can be seen that the macroinitiator had a stable helical structure. The copolymerization with D-1 was studied. As Fig. 4a shown, the molar circular dichroism of the block polymer decreased with increasing Mn, which indicated that the poly-D-1 block had a destructive effect on the helix structure of the copolymer. As for the approximate linear relationship in Fig. 4b, the reason may be that the number of the repeating unit increased (increased UV absorption) while the helical structure of the copolymer changed (decreased optical activity) simultaneously.

|
Download:
|
| Fig. 4. (a) Molar circular dichroism and UV-vis spectra of poly(L-150-b-D-1n) measured in THF at 298 K. (b) The plot of molar circular dichroism of poly(L-150-b-D-1n) measured in THF at 298 K as a function of Mn (c = 0.03 g/L). | |
{kind=link}
The process of block polymerization of poly(L-150-b-rac-150) illustrated that the new Ni complex can achieve the enantiomer-selectivity. Sampling from copolymerization system of poly (L-150-b-rac-150) to analyze the obtained copolymer and recovered monomer. A large amount of methanol was added to quench the sample. The formed precipitate was isolated by centrifugation to afford copolymer and supernatant. The ee values of the supernatant were determined by high performance liquid chromatography (HPLC) using columns with chiral stationary phase. As shown in Fig. 5a, with the conversion of rac-1, the ee value continuously increased and finally reached the maximum value of 34%. It was confirmed that this polymerization was a kind of kinetic resolution, in which L-1 was preferentially polymerized over the antipode. For copolymers, the correspondence between the molar circular dichroism and Mn was shown in Fig. 5b, from which it can be observed that the change of molar circular dichroism was slightly in the first stage of polymerization. Combined with the kinetic curve, the reason may be that L-1 preferentially polymerized over the antipode D-1 at the same concentration. When the Mn exceeded 28.1 kDa, the molar circular dichroism changed rapidly. This is due to the increase of relative concentration of D-1 which made the advantages of L-1 weakened. The change in concentration still could not offset the difference in kL-1 and kD-1. Therefore, the ee value was continuously increased throughout the process. Meanwhile, there was still a considerable CD intensityat the end of the copolymerization.

|
Download:
|
| Fig. 5. (a) Plot of ee value of the supernatant as a function of conversion of rac-1 during the copolymerization of poly(L-150-b-rac-150). (b) Plot of molar circular dichroism of poly(L-150-b-rac-1o) sampled during the copolymerization process measured in THF at 298 K as a function of Mn (c = 0.03 g/L). | |
{kind=link}
In summary, we have demonstrated the enantiomer-selective polymerization of chiral allene derivative monomer by using chiral phosphine allylnickel(Ⅱ) complexcatalyst.The polymerization rate of L-1 was 1.9 times that of D-1. During the polymerization of poly (L-150-b-rac-150), both the obtained copolymer and the recovered monomer exhibited optical activity. It illustrated that the subtle change in the metal coordination environment had a huge impact on the polymerization. The chiral environment from the ligand was effective for the enantiomeric-selectivity. We believe that higher selectivity will be achieved by matching the monomers with new ligand of the chimeric center.
AcknowledgmentsThis work was sponsored by the National Natural Science Foundation ofChina (Nos. 21622402, 51673057, and 21574036). Z. Q. Wu thanks the Thousand Young Talents Program of China and the Open Project of State Key Laboratory of Supramolecular Structure and Materials (No. sklssm201624) for Financial Support.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/jxclet.2018.03.002.
| [1] |
S. Nilsson, B. Jonsson, L. Piculell, Macromolecules 22 (1989) 2367-2375. DOI:10.1021/ma00195a063 |
| [2] |
L. Pauling, R.B. Corey, H.R. Branson, Proc. Natl. Acad. Sci. USA 37 (1951) 205-211. DOI:10.1073/pnas.37.4.205 |
| [3] |
J.D. Watson, F.H.C. Crick, Nature 171 (1953) 737-738. DOI:10.1038/171737a0 |
| [4] |
E. Yashima, K. Maeda, H. Iida, et al., Chem. Rev. 109 (2009) 6102-6211. DOI:10.1021/cr900162q |
| [5] |
S.W. Smith, Toxicological Sciences 110 (2009) 4-30. DOI:10.1093/toxsci/kfp097 |
| [6] |
J.Y. Wang, F. Zhao, M. Zhang, et al., Chin. Chem. Lett. 19 (2008) 1248-1251. DOI:10.1016/j.cclet.2008.07.003 |
| [7] |
N. Hoshikawa, Y. Hotta, Y. Okamoto, J. Am. Chem. Soc. 125 (2003) 12380-12381. DOI:10.1021/ja035871y |
| [8] |
M. Teraguchi, K. Mottate, S.Y. Kim, et al., Macromolecules 38 (2005) 6367-6373. DOI:10.1021/ma050089z |
| [9] |
L. Zhou, B.F. Chu, X.Y. Xu, et al., ACS Macro. Lett. 6 (2017) 824-829. DOI:10.1021/acsmacrolett.7b00439 |
| [10] |
L. Yang, Y. Tang, N. Liu, et al., Macromolecules 49 (2016) 7692-7702. DOI:10.1021/acs.macromol.6b01870 |
| [11] |
B. Chen, J.P. Deng, X. Cui, W.T. Yang, Macromolecules 44 (2011) 7109-7114. DOI:10.1021/ma2013628 |
| [12] |
M. Tsuji, R. Sakai, T. Satoh, et al., Macromolecules 35 (2002) 8255-8257. DOI:10.1021/ma020861n |
| [13] |
S.W. Seidel, T.J. Deming, Macromolecules 36 (2003) 969-972. DOI:10.1021/ma025844c |
| [14] |
Y. Okamoto, T. Nakano, Chem. Rev. 94 (1994) 349-372. DOI:10.1021/cr00026a004 |
| [15] |
Z.Q. Wu, K. Nagai, M. Banno, et al., J. Am. Chem. Soc. 131 (2009) 6708-6718. DOI:10.1021/ja900036n |
| [16] |
T. Endo, I. Tomita, Prog. Polym. Sci. 22 (1997) 565-600. DOI:10.1016/S0079-6700(96)00022-6 |
| [17] |
G. Lu, Y. Li, H. Gao, et al., J. Polym. Sci. Part A:Polym. Chem. 51 (2013) 1099-1106. DOI:10.1002/pola.26470 |
| [18] |
K. Mochizuki, I. Tomita, Macromolecules 39 (2006) 6336-6340. DOI:10.1021/ma052374o |
| [19] |
J. Wang, I. Tomita, Macromolecules 34 (2001) 4294-4295. DOI:10.1021/ma001344c |
| [20] |
T. Kino, M. Taguchi, A. Tazawa, I. Tomita, Macromolecules 39 (2006) 7474-7478. DOI:10.1021/ma052492b |
| [21] |
I. Tomita, Y. Kondo, K. Takagi, T. Endo, Macromolecules 27 (1994) 4413-4414. DOI:10.1021/ma00093a052 |
| [22] |
K. Takag, I. Tomita, T. Endo, Macromolecules 31 (1998) 6741-6747. DOI:10.1021/ma9800921 |
| [23] |
A. Narumi, R. Sakai, S. Ishido, et al., Macromolecules 40 (2007) 9272-9278. DOI:10.1021/ma071162j |
| [24] |
T. Nishimura, Y. Ichikawa, T. Hayashi, et al., Organometallics 28 (2009) 4890-4893. DOI:10.1021/om9003797 |
| [25] |
J.J. Cheng, T.J. Deming, Macromolecules 32 (1999) 4745-4747. DOI:10.1021/ma990241v |
| [26] |
Y.Y. Zhu, T.T. Yin, X.L. Li, et al., Macromolecules 47 (2014) 7021-7029. DOI:10.1021/ma5019022 |