 2019, Vol. 30
2019, Vol. 30 


Polymer blending has received much attention to develop new materials with combined properties of each component [1-3]. However, most polymer blends are thermodynamically immiscible due to the high molecular weight and unfavorable interaction between different polymers, which leads to deteriorating performances by simple blending. Effective compatibilization for polymer blends is thus the prerequisite to obtain polymeric materials with satisfactory properties. Two approaches are generally adopted to realize compatibilization: One is to add premade block or graft copolymers during blending of immiscible polymers [4-12], and the other is to form in-situ block or graft copolymers at the interfacial regions, which is called reactive compatibilization [13-20].
Over the past few decades, benefited from the progress of compatibilization technology, substantial polymer blends with prominent performances have been developed and applied to all areas of our daily life. As an example, polycarbonate/acrylonitrile-butadiene-styrene copolymer (PC/ABS) blend is one of the most successful commercial polymer blends which has been widely used in automotive and electronic areas [3, 21-24]. Nevertheless, there are several shortcomings of PC/ABS blends, such as high cost of PC and poor solvent resistance. As a replacement of PC/ABS, polyamide (PA)/ABS blends, characteristic of low cost, superior chemical resistance, processability, dimensional stability, as well as balanced mechanical properties, have attracted considerable interest recently from both academia and industry [25-28]. Appropriate compatibilization is still necessary due to inherent immiscibility between PA and ABS.
Several reactive compatibilization strategies have been reported for PA/ABS blends in literatures. One approach is to incorporate a functional block or graft copolymer capable of reacting with the amino groups in PA and being miscible with ABS phases, such as styrene-maleic anhydride copolymer (SMA) [29], imidized acrylic polymer (IA) [30], styrene-acrylonitrile-maleic anhydride terpolymer (SANMA) [26] and glycidyl methacrylate-methyl methacrylate copolymer (GMA-MMA) [1]. There are several investigations on compatibilizing PA/ABS blends by a poly(maleic anhydride)-grafted ABS or poly(glycidyl methacrylate)-grafted ABS, instead of adding a third component as a compatibilizer [31-33]. However, most of thoseworksmainly focuson themorphologyof the PA/ABSblendsor involve in an attempt of toughening at the expense of tensile properties. It is still a longstanding challenge to simultaneously enhancethe strengthand toughnessof thePA/ABSblends, whichisof very important practical significance to create a new set of engineering materials.
In recent years, our group has developed a series of effective multiphase compatibilizers using MAH and St dual monomers in a meltgrafting strategy [34-40]. We have successfully obtained super-tough and strong PA-based ternary and quaternary blends [41-43]. In the current study, ABS and poly[styrene-b-(ethylene-co-butylene)-b-styrene] (SEBS) aremelt-graftedwith twomonomersMAH and St to obtain the multi-phase compatibilizers ABS-g-(MAH-co-St) and SEBS-g-(MAH-co-St) and then theyare utilized to compatibilize PA/ABS blends. The compatibilizers are adopted because MAH can react with the terminal amino group in PA, and the graft degree of functional monomer can be increased in the presence of St, which has been demonstrated in our previous work [35]. The elastomer SEBS plays a key role in toughening the blends, which will be elaborated in detail in this communication. The morphologies and mechanical properties of these blends with changing combination of the compatibilizers will be systematically investigated, and their correlation will be elucidated in detail. These blends are promising candidates or replacements for PC/ABS blends.
The multi-monomer melt-grafted compatibilizers g-ABS and g-SEBS were prepared with a twin-screw extruder (Φ = 26 mm, L/D = 40), where g-ABS and g-SEBS refer to ABS-g-(MAH-co-St) and SEBS-g-(MAH-co-St), respectively, if otherwise not specified in the text. For a typical grafting process, ABS or SEBS pellets, MAH, St and initiator dicumyl peroxide (DCP) were premixed before charged into the extruder. The concentrations of MAH, St and DCP were 3 wt%, 3 wt%, and 0.1 wt%, respectively, to ABS or SEBS. The temperature profile of the barrel was set at 170-210 ℃, and the rotation speed of the screws was 80 rpm. The extrudates were pelletized, and then dried in a vacuum oven at 60 ℃ for 24 h. The grafting ratios of MAH in g-ABS and g-SEBS are 2.34 wt% and 2.28 wt% determined by a back-titration procedure, and the details of the titration procedure can be found in the literature [44]. A series of PA6/ABS blends were prepared via melt blending using a twin-screw extruder (Φ = 26 mm, L/D = 40). The temperature file of the barrel was 200-230 ℃ from hopper to die with a rotation speed of 80 rpm. The morphologies of the blends were investigated using field emission scanning electron microscopy (FESEM) (JSM-7401, JEOL Instrument), and atomic force microscopy (AFM) (SPM-9700, Shimadzu). The tensile and impact tests were performed by a GOTECH-2000 universal testing machine and an XJUD-5.5 impact testing machine, respectively.
Fig. 1 shows the FTIR spectra of the compatibilizers g-SEBS and g-ABS as well as the two neat polymers. As shown in Fig. 1a, the new absorption bands at 1781 and 1863 cm-1 for g-SEBS are assigned to the stretching vibration of the carbonyl groups in the grafted anhydride unit [35, 40, 44]. The characteristic absorption bands are also identified in the g-ABS sample shown in Fig. 1b. These results imply that the MAH monomers are successfully grafted onto SEBS and ABS molecules.

|
Download:
|
| Fig. 1. FTIR spectra of neat SEBS (a) and ABS (b), and the prepared compatibilizers. | |
Fig. 2 shows the FESEM micrographs of the PA6/ABS (70/30) binary blends and compatibilized ternary blends after cryofractured in liquid nitrogen. PA6 and ABS are highly immiscible and thus form very large dispersed domains (over 8 μm) and sharp interfaces, as shown in Figs. 2a and a'. The dispersed ABS domains can be easily removed from the PA6 matrix due to poor interfacial adhesion between the two immiscible polymer pairs. When 15 wt% ABS is replaced by g-ABS, the size of the dispersed phase is dramatically decreased as shown in Figs. 2b and b'. It should be noted that most particles of the minor phase are intercalated in the matrix and are broken apart at the cryo-fractured surface (as marked by red circle in the inset of Fig. 2b), which indicates the enhanced interfacial interaction between the matrix and the dispersed phase in the compatibilized blend. The significant change of the morphology implies the significant improvement on the compatibility between PA6 and ABS. which results from the in-situ reaction between the g-ABS and the PA6.

|
Download:
|
| Fig. 2. FESEM micrographs of (a) and (a') PA6/ABS (70/30), (b) and (b') PA6/ABS/g-ABS (70/15/15), (c) and (c') PA6/ABS/g-SEBS (70/15/15), and (d) and (d') PA6/g-ABS/g-SEBS (70/15/15), respectively. In (a') and (b'), the ABS phase was extracted by tetrahydrofuran while in (c') and (d'), the SEBS phase was extracted by cyclohexane. The insets show the morphologies at a higher magnification. | |
In order to impart superior toughness to PA6/ABS blends, the compatibilizer in PA6/ABS/g-ABS (70/15/15) blend is substituted by an elastomer-based compatibilizer, g-SEBS. Subsequently, both of the two compatibilizers mentioned above are used to further compatibilize the PA6/ABS blends. Figs. 2c and d reveal the morphologies of the cryo-fractured surfaces of PA6/ABS/g-SEBS (70/15/15) ternary blend and PA6/g-ABS/g-SEBS (70/15/15) ternary blend. As seen in Fig. 2c, the dispersed domain size is much smaller than the neat PA6/ABS (70/30) binary blend, which indicates that the g-SEBS effectively decreases the size of dispersed phase as g-ABS does. The detailed morphologies of the dispersed phase can be observed after selective etching the SEBS component by cyclohexane, which keeps ABS phase intact. It is noteworthy that both ABS and SEBS phases will be extracted if tetrahydrofuran is still used to etch the PA6/ABS/g-SEBS and PA6/g-ABS/g-SEBS ternary blends. As shown in Fig. 2c', the dispersed phases actually form a core-shell structure, where ABS phase is the core and SEBS is the shell. A considerable amount of SEBS domains also exist as discrete particles in PA6 matrix with an average diameter less than 0.5 μm. In this case, the SEBS phase locates at the interfacial regions and even distributes in PA6 matrix individually because of the in-situ reaction between g-SEBS and PA6. Fig. 3c is a schematic description of the core-shell structure comprised of SEBS and ABS phases, together with the separate SEBS domains. With incorporation of both g-ABS and g-SEBS, as found in Figs. 2d and d', the average size of the core-shell domains in the ternary blend is significantly reduced and the interfaces between dispersed domains and the matrix become vague, which implies a further improvement in the compatibility among different components. The schematic illustration is shown in Fig. 3d.

|
Download:
|
| Fig. 3. (a) and (a') AFM micrographs of PA6/ABS/g-SEBS (70/15/15); (b) and (b') AFM micrographs of PA6/g-ABS/g-SEBS (70/15/15), in which (a) and (b) are height images, while (a') and (b') are adhesion mapping images. (c) and (d) schematic diagram of morphology PA6/ABS/g-SEBS (70/15/15) and PA6/g-ABS/g-SEBS (70/15/15), respectively. | |
AFM with mapping function was also carried out to study the micro-phase distribution, and the micrographs are presented in Fig. 3. The dispersed phases can be hardly distinguished from the matrix in the height images (Figs. 3a and b). However, the detailed distribution of the dispersed phase can be highlighted in the adhesion mapping images, which convey the information of viscoelasticity or modulus of different constituents in the blends. As shown in Figs. 3a' and b', the bright regions represent the soft component SEBS with lower modulus, while the dark regions represent the hard PA6 and ABS. It can be clearly observed that the minor components are dispersed as finer domains in the PA6/g-ABS/g-SEBS (70/15/15) ternary blend than the PA6/ABS/g-SEBS (70/15/15). Through FESEM and AFM observation, the morphologies of the two blends are consistent with the schematic description in Figs. 3c and d. In PA6/ABS/g-SEBS (70/15/15) blend (Fig. 3c), the ABS phase is dispersed as droplets encapsulated by the g-SEBS phase to form a core-shell structure in PA6 matrix due to the reactivity between PA6 and MAH in g-SEBS. Some g-SEBS can be also dispersed in the matrix individually as mentioned before. When both g-ABS and g-SEBS are added into the blend (Fig. 3d), more of the minor phases will be dispersed separately in the matrix driven by the chemical reaction. Meanwhile, there are also some ABS droplets encapsulated by SEBS because of the higher melt viscosity of ABS compared with SEBS.
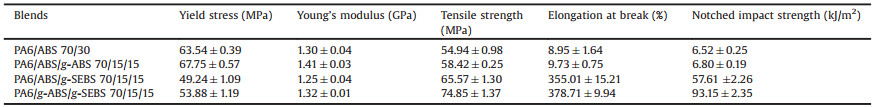
The stress-strain curves and notched impact strength of different PA6/ABS blends are depicted in Fig. 4 and the corresponding data of tensile and impact properties are listed in Table 1. Compared with the PA6/ABS (70/30) blend, the incorporation of 15 wt% g-ABS in the PA6/ABS/g-ABS (70/15/15) slightly increases the yield stress and Young's modulus of the binary blend due to the enhancement of the interfacial interaction between the matrix and ABS. However, the elongations of both blends are very low, which indicates the poor toughness of these binary blends due to the inherent rigidity of the two components. Therefore, the elastomer-based compatibilizer g-SEBS is used to improve the toughness of the blends. As expected, when the 15 wt% of g-ABS is replaced by the g-SEBS, the elongation at break of the PA6/ABS/g-SEBS (70/15/15) ternary blend is increased significantly, despite of a decline in yield stress and modulus. When the two compatibilizers are both added into the blend, there is a comprehensive improvement in yield stress, Young's modulus, elongation at break and tensile strength, compared with PA6/ABS/g-SEBS (70/15/15) blend. The remarkable reinforcement effect of the compatibilizers can be attributed to the enhanced interfacial adhesion due to the in-situ formation of the grafted copolymer between compatibilizers and PA6 during the melt blending procedure. When subjected to tensile stress, the matrix can sustain a steady plastic deformation for the compatibilized blends, rather than a premature failure due to stress concentration at defective interfaces. Accordingly, the necking process can develop steadily, so that the strain hardening can be realized subsequently to reach a higher elongation and tensile strength. The increased interaction among the components contributes to an improvement in stiffness of the blend, and thus leads to a higher yield stress and Young's modulus for the PA6/ABS binary blend (or the PA6/ABS/SEBS ternary blend).

|
Download:
|
| Fig. 4. (a) The stress-strain curves and (b) notched impact strength of PA6/ABS binary and ternary blends. | |
|
|
Table 1 Mechanical properties of PA6/ABS binary and ternary blends. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fig. 4b reveals the effect of compatibilizers on the impact performance of the PA6/ABS binary and ternary blends. The replacement of ABS with the compatibilizer g-ABS has little effect on the impact property of PA6/ABS blends due to lack of effective energy dissipation mechanism in the brittle binary blends. As a contrast, the brittle-ductile transition occurs with the incorporation of g-SEBS. The notched impact strength of the PA6/ABS/g-SEBS (70/15/15) ternary blend has improved by 780% in comparison with PA6/ABS (70/30) binary blend. When both the two compatilizers are added into the blend, the notched impact strength of the PA6/g-ABS/g-SEBS (70/15/15) ternary blend is further increased by 60% compared with PA6/ABS/g-SEBS (70/15/15) blend, or 1300% compared with PA6/ABS (70/30) blend. It is noteworthy that the tensile properties of PA6/g-ABS/g-SEBS (70/15/15) blend, namely yield stress, Young's modulus, elongation at break and tensile strength, are all better than PA6/ABS/g-SEBS (70/15/15) blend, which are quite different from the general toughening mechanism at the severe expense of stiffness and tensile strength. The decreased domain size and the formation of encapsulation structure account for the efficient toughening effect of g-ABS and g-SEBS. Grafting MAH onto SEBS facilitates the rubber phases to encapsulate ABS domains, which has been confirmed by SEM. The resulted "soft shell-encapsulating-hard core" structure is more effective for toughening of the blends [43]. The soft shell can initiate massive crazes and shear bands at the interfaces between minor phases and the matrix to dissipate the impact energy [45]. The increased interfacial adhesion due to the in-situ reaction between MAH and amino groups transfers the energy to the rubber phase without causing the interface to develop into large cracks. In addition, the significantly decreased particle size is a crucial reason for the significant improvement of the impact performance. On one hand, the reduced size of dispersed phases means the increase of interface area, which can dissipate more impact energy. On the other hand, more and more dispersed domains smaller than 0.5 mm emerges in the matrix, which falls in the optimal range of particle diameter, namely 0.2-0.4 μm, for toughening nylon blends [46, 47]. Therefore, the multi-phase compatibilizers g-SEBS and g-ABS show excellent synergistic effect on simultaneously toughening and reinforcing the ternary blends, and there is a good correspondence between the mechanical properties and the micro-structure of the blends.
In summary, we have developed an effective strategy for compatibilization of PA/ABS blends using multi-phase compatibilizers, g-ABS and g-SEBS. The incorporation of g-ABS drastically reduces the dispersed domain size of ABS, and g-SEBS creates a "soft shell" to encapsulate the "hard core" of ABS. The fine dispersion and the unique "core-shell" structure of PA6/g-ABS/g-SEBS (70/15/15) lead to synergistic improvements in tensile strength and impact performance. Compared with PA6/ABS (70/30) blend, the tensile strength and notched impact strength of PA6/g-ABS/g-SEBS (70/15/15) blend are increased by 36% and 1300%, respectively. Our work presents a facile and efficient way to obtain PA6/ABS blends with high performances, which exhibit a promising potential in the automotive and electronic areas.
AcknowledgmentWe express our sincere gratitude to the National Natural Science Foundation of China (No. 51633003) for the financial support.
| [1] |
R.A. Kudva, H. Keskkula, D.R. Paul, Polymer 39 (1998) 2447-2460. DOI:10.1016/S0032-3861(97)00583-1 |
| [2] |
D.R. Paul, C.B. Bucknall, Polymer Blends:Formulation and Performance. New York: Wiley, 2000.
|
| [3] |
L.A. Utracki, Polymer Alloys and Blends:Thermodynamics and Rheology. Munich: Hanser, 1989.
|
| [4] |
Y. Xu, C.M. Thurber, C.W. Macosko, T.P. Lodge, M.A. Hillmyer, Ind. Eng. Chem. Res. 53 (2014) 4718-4725. DOI:10.1021/ie4043196 |
| [5] |
M. Maric, C.W. Macosko, J. Polym. Sci. Part B Polym. Phys. 40 (2002) 346-357. DOI:10.1002/(ISSN)1099-0488 |
| [6] |
A. Adedeji, S. Lyu, C.W. Macosko, Macromolecules 34 (2001) 8663-8668. DOI:10.1021/ma001944b |
| [7] |
C.W. Macosko, P. Guegan, A.K. Khandpur, et al., Macromolecules 29 (1996) 5590-5598. DOI:10.1021/ma9602482 |
| [8] |
X.M. Xie, Y. Chen, Z.M. Zhang, et al., Macromolecules 32 (1999) 4424-4429. DOI:10.1021/ma9805046 |
| [9] |
J. Kim, R.W. Sandoval, C.M. Dettmer, S.T. Nguyen, J.M. Torkelson, Polymer 49 (2008) 2686-2697. DOI:10.1016/j.polymer.2008.04.008 |
| [10] |
R. Bahrami, T.I. Lobling, H. Schmalz, A.H.E. Muller, V. Altstadt, Polymer 80 (2015) 52-63. DOI:10.1016/j.polymer.2015.10.039 |
| [11] |
R. Bahrami, T.I. Lobling, H. Schmalz, A.H.E. Muller, V. Altstadt, Polymer 109 (2017) 229-237. DOI:10.1016/j.polymer.2016.12.044 |
| [12] |
A. Bharati, R. Cardinaels, T. Van der Donck, et al., Polymer 108 (2017) 483-492. DOI:10.1016/j.polymer.2016.12.015 |
| [13] |
H. Koriyama, H.T. Oyama, T. Ougizawa, et al., Polymer 40 (1999) 6381-6393. DOI:10.1016/S0032-3861(98)00856-8 |
| [14] |
J.B. Zhang, T.P. Lodge, C.W. Macosko, Macromolecules 38 (2005) 6586-6591. DOI:10.1021/ma050530l |
| [15] |
M. Harada, K. Iida, K. Okamoto, H. Hayashi, K. Hirano, Polym. Eng. Sci. 48 (2008) 1359-1368. DOI:10.1002/(ISSN)1548-2634 |
| [16] |
G.C. Liu, Y.S. He, J.B. Zeng, Q.T. Li, Y.Z. Wang, Biomacromolecules 15 (2014) 4260-4271. DOI:10.1021/bm5012739 |
| [17] |
C.M. Thurber, Y.W. Xu, J.C. Myers, T.P. Lodge, C.W. Macosko, ACS Macro Lett. 4 (2015) 30-33. DOI:10.1021/mz500770y |
| [18] |
H.T. Wang, W.Y. Dong, Y.J. Li, ACS Macro Lett. 4 (2015) 1398-1403. DOI:10.1021/acsmacrolett.5b00763 |
| [19] |
A.M. Zolali, B.D. Favis, Polymer 114 (2017) 277-288. DOI:10.1016/j.polymer.2017.02.093 |
| [20] |
A.D. Todd, R.J. McEneany, V.A. Topolkaraev, C.W. Macosko, M.A. Hillmyer, Macromolecules 49 (2016) 8988-8994. DOI:10.1021/acs.macromol.6b02080 |
| [21] |
R. Greco, A. Sorrentino, Adv. Polym. Technol. 13 (1994) 249-258. DOI:10.1002/adv.1994.060130401 |
| [22] |
D.R. Paul, J.W. Barlow, J. Macromol. Sci. Rev. Macromol. Chem. Phys. C 18 (1980) 109-168. DOI:10.1080/00222358008080917 |
| [23] |
G.S. Wildes, T. Harada, H. Keskkula, et al., Polymer 40 (1999) 3069-3082. DOI:10.1016/S0032-3861(98)00521-7 |
| [24] |
M.P. Lee, A. Hiltner, E. Baer, Polymer 33 (1992) 685-697. DOI:10.1016/0032-3861(92)90323-O |
| [25] |
Y.J. Li, H. Shimizu, Macromol. Rapid Commun. 26 (2005) 710-715. DOI:10.1002/(ISSN)1521-3927 |
| [26] |
R.A. Kudva, H. Keskkula, D.R. Paul, Polymer 41 (2000) 239-258. DOI:10.1016/S0032-3861(99)00106-8 |
| [27] |
R.A. Kudva, H. Keskkula, D.R. Paul, Polymer 41 (2000) 225-237. DOI:10.1016/S0032-3861(99)00105-6 |
| [28] |
X.Q. Liu, W. Yang, B.H. Xie, M.B. Yang, Mater. Des. 34 (2012) 355-362. DOI:10.1016/j.matdes.2011.08.028 |
| [29] |
B.K. Kim, Y.M. Lee, H.M. Jeong, Polymer 34 (1993) 2075-2080. DOI:10.1016/0032-3861(93)90734-R |
| [30] |
B. Majumdar, H. Keskkula, D.R. Paul, Polymer 35 (1994) 5453-5467. DOI:10.1016/S0032-3861(05)80009-6 |
| [31] |
Y. Fu, H. Song, C. Zhou, H. Zhang, S. Sun, Polym. Bull. 70 (2013) 1853-1862. DOI:10.1007/s00289-012-0879-7 |
| [32] |
S. Shulin, C. Zhuo, S. Lili, Z. Huixuan, J. Appl. Polym. Sci. 121 (2011) 909-915. DOI:10.1002/app.33648 |
| [33] |
S. Sun, Z. Chen, H. Zhang, Polym. Bull. 61 (2008) 443-452. DOI:10.1007/s00289-008-0971-1 |
| [34] |
X.M. Xie, N.H. Chen, B.H. Guo, S. Li, Polym. Int. 49 (2000) 1677-1683. DOI:10.1002/(ISSN)1097-0126 |
| [35] |
Y. Li, X.M. Xie, B.H. Guo, Polymer 42 (2001) 3419-3425. DOI:10.1016/S0032-3861(00)00767-9 |
| [36] |
X.M. Xie, Y. Li, J.C. Zhang, X. Yang, Acta Polym. Sin. 33 (2002) 7-12. |
| [37] |
D. Wang, X.M. Xie, Polymer 47 (2006) 7859-7863. DOI:10.1016/j.polymer.2006.09.026 |
| [38] |
Y. Li, D. Wang, J.M. Zhang, X.M. Xie, J. Appl. Polym. Sci. 119 (2011) 1652-1658. DOI:10.1002/app.v119:3 |
| [39] |
Y. Li, D. Wang, J.M. Zhang, X.M. Xie, Polym. Bull. 66 (2011) 841-852. DOI:10.1007/s00289-010-0380-0 |
| [40] |
D. Wang, Y. Li, X.M. Xie, B.H. Guo, Polymer 52 (2011) 191-200. DOI:10.1016/j.polymer.2010.11.019 |
| [41] |
H. Li, X. Sui, X.M. Xie, Polymer 123 (2017) 240-246. DOI:10.1016/j.polymer.2017.07.024 |
| [42] |
H. Li, X.M. Xie, Polymer 108 (2017) 1-10. DOI:10.1016/j.polymer.2016.11.044 |
| [43] |
H. Li, X. Sui, X.M. Xie, Chin. J. Polym Sci. 36 (2018) 848-858. DOI:10.1007/s10118-018-2102-2 |
| [44] |
C. Li, Y. Zhang, Y. Zhang, Polym. Test. 22 (2003) 191-195. DOI:10.1016/S0142-9418(02)00079-X |
| [45] |
W.C. Shi, G.H. Fredrickson, E.J. Kramer, et al., ACS Nano 10 (2016) 2054-2062. DOI:10.1021/acsnano.5b06215 |
| [46] |
C.B. Bucknall, D.R. Paul, Polymer 50 (2009) 5539-5548. DOI:10.1016/j.polymer.2009.09.059 |
| [47] |
C.B. Bucknall, D.R. Paul, Polymer 54 (2013) 320-329. DOI:10.1016/j.polymer.2012.11.019 |