 2019, Vol. 30
2019, Vol. 30 


b Department of Chemistry, Beijing Normal University, Beijing 100050, China
Mechanically interlocked molecular architectures such as catenanes [1-9], rotaxanes [1-3, 6-8, 10], molecular knots [5, 11, 12] and molecular borromean rings [13-16], have been extensively explored in different fields. Among all of them, rotaxanes have captured the imaginations of a wide spectrum of chemists as a result of not only their undisputed structural beauty but also their potential applications in sensors [17-24], catalysts [25-28] and functional materials [29-32]. The past twenty years have witnessed the explosion of developing novel rotaxanes with different properties [11, 33-35]. However, although various rotaxanes have been reported, most of them focused on the construction of [2]rotaxanes [36-41]. There have been a few examples of the higher ordered rotaxanes [42-47]. The construction of highly ordered and discrete multirotaxanes with the definite shapes still remains a challenge for chemists mainly in consequence of the synthetic difficulty. We have recently reported the successful construction of discrete stimuli-responsive multirotaxans with well-defined supramolecular metallacycles as cores via coordination-driven self-assembly [48, 49]. Inspired by our previous progress on highly ordered multirotaxanes [50], we envisioned that the introduction of porphyrin into the rotaxanes would endow the resulting multirotaxans with not only the well-defined symmetrical structure but also interesting self-assembly behaviour in solution [51]. Notably, the study of self-assembly behaviour of multirotaxans could help to better understand their applications on surface.
Herein, we designed and synthesised a highly symmetric [5]rotaxane with zinc porphyrin as core and platinum-acetylide moiety, trans-Pt(PEt3)2(C≡CR')2 as linkers in which dipropylpillar[5]arene (DPP[5]A) was locked by the block substituted-phenoxyl on every branch (Fig. 1). The synthetic strategy involved the highyield coupling reaction between the [2]rotaxane precursor 3 with platinum-iodine as terminal and the alkyne core to form the bridged platinum-acetylide as reported [49]. The [5]rotaxane 1 was well characterized with NMR spectroscopy and MALDI-TOF mass spectrometry. UV–vis and emission spectroscopy indicated J-type aggregation of the [5]rotaxane in solution with the help of π–π interaction as indicated by the concentration-dependent 1H NMR experiments. Furthermore, the [5]rotaxane could selfassembly into nanospheres or nanofibers depending on the polarity of the solvents. Notably, the model complex without pillarene displayed the unordered morphology at the same condition, thus suggesting that the existence of rotaxanes endowed the complex with a relative rigid structure to facilitate the formation of the ordered aggregates. Moreover, an evolution from nanofibers to nanospheres caused by the twist of nanofibers was observed for the [5]rotaxane in polar solution.

|
Download:
|
| Fig. 1. Cartoon representation of [5]rotaxane 1. | |
{kind=link}
The synthetic route of branched [5]rotaxane 1 is outlined in Scheme S1 (Supporting information). The reported compound, Zn (Ⅱ) tetra(4-ethynylphenyl)porphyrin 11 [52], was used as one of the key intermediates because of its exclusive reaction sites with platinum acetylide species. Another key intermediate was the [2]rotaxane 3 [49] with platinum acetylide in terminal. The coupling reaction of these two key intermediates, 11 and 3, in the presence of CuI as catalyst resulted in the final product [5]rotaxane 1 in high yield (86%).
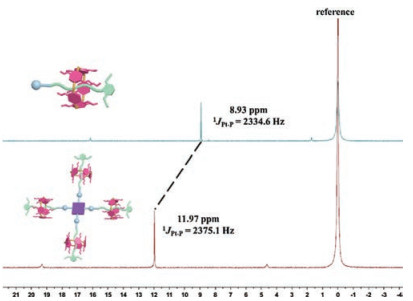
The molecular structure of the obtained branched [5]rotaxane was well characterized with multiple nuclear NMR (1H, 13C, 31P and 1H–1H COSY) spectroscopy and mass MALDI-TOF mass spectrometry (Fig. 2, Figs. S1, S38 and S40 in Supporting information). The 1H NMR spectrum of 1 (Fig. S38a) exhibited only one signal in aromatic region for zinc porphyrin core, suggesting the reserving of D4h symmetry. Moreover, the methylene peaks of the alkane chain splitted into six sets of signals and shifted up-field obviously even up to -2.2 ppm, which was attributed to the shielding effects of the benzene on the DPP[5]A. This feature was consistent with the description of the similar rotaxane structure based on pillar[5]arene and alkane chains [49, 50], thus strongly supporting the existence of rotaxane moieties in the final branched [5]rotaxane. Furthermore, NOE correlation signals were observed between the methyl protons H16 on DPP[5]A and protons H12 and H13, indicating that the alkyl chain was deeply threaded into the DPP[5]A cavity (Fig. S41 in Supporting information). The 31P NMR spectrum (Fig. 2) revealed a single and sharp peak at ca. 11.0 ppm shifted downfield from the precursor 3 by ca. 3.0 ppm due to the electron withdrawal of the large π-conjugated bridges. With the considerable effort, high quality mass signal of 1 was observed in the MALDI-TOF mass spectrum (Fig. S1). A peak at 8251.289 (m/z) was observed, which was consistent with the M with a deviation of less than 0.45 Da. In all words, all of these results provided strong support for the formation of the desired highly symmetric [5]rotaxane structure.

|
Download:
|
| Fig. 2. 31P NMR spectra (161.9 MHz, 298 K) of the precursor 3 (top) and [5]rotaxane 1 (bottom) in CD2Cl2. | |
{kind=link}
Though many efforts have been made to grow X-ray quality single crystals of the [5]rotaxane 1, all attempts have been unsuccessful so far. To gain further insight into the structural characteristics of 1, the simulation by PM6 semiempirical molecular orbital method was conducted. The optimized result indicated that the zinc porphyrin core reserved the planar conformation with four branched rotaxane arms in a length of ca. 35 Å around it (Fig. S1). Moreover, the size of the whole molecule in vacuum was determined by ca. 56 Å × 52 Å.
The photophysical properties at the single molecule level were investigated in detail by using UV–vis and emission spectrometry. The ground state absorption spectra of the [5]rotaxane in dichlormethane (5.0 × 10-6 mol/L) were shown in Fig. S3 in Supporting information. The absorption spectrum exhibited the characteristic signals of the zinc porphyrin core, the Soret band and Q band. The Q band displayed two resolved absorptions at 553 nm and 595 nm, indicating the maintenance of symmetry after the coupling reaction. The higher energy band at 295 nm was derived from π–π* transitions of the triethynylbenzene-bridges whereas the absorption at 344 nm was attributable to the MLCT band of platinum-acetylides [53].
When the [5]rotaxane was excited at the MLCT band of platinum-acetylide units (344 nm), two emission peaks at 611 nm and 460 nm were observed. Upon excitation at the Soret band (435 nm), only one emission peak at 611 nm was found, which was the characteristic of zinc porphyrin nucleus with the energy transfer from platinum-acetylide moiety to the zinc porphyrin core. A mixture of compounds 11 and 3 in a ratio of 1:4 displayed weak emission from the porphyrin but strong signal from platinum-acetylide units upon excitation at 344 nm, which was consistent with the intramolecular energy transfer.
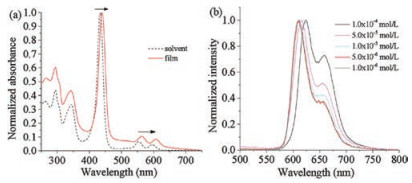
Moreover, the aggregation behaviour of 1 both in solution and on the surface was investigated (Fig. 3 and Fig. S3). When the sample was deposited onto a quartz substrate as a film, both the Soret and Q bands of the zinc porphyrin core were slightly redshifted (6.0 nm for the Soret band, 8.0 nm and 13 nm for the Q band, respectively) and significantly broadened when compared to that in dichloromethane (5.0 × 10-5 mol/L) (Fig. 3a). Moreover, upon warming the solution from 10 ℃ to 40 ℃, the Soret band showed slight hypsochromic shift (Fig. S3a). All these findings suggested the J-type aggregation process, which might be promoted by the intermolecular π–π interaction in cooperation with the hydrophobic interactions [51].

|
Download:
|
| Fig. 3. (a) Normalized UV–vis spectra of 1 in dichloromethane (5.0 × 10-6 mol/L) and in the film state; (b) emission spectra of 1 in dichloromethane at differentconcentrations (λex = 435 nm). (For interpretation of the references to colour in the text, the reader is referred to the web version of this article.) | |
{kind=link}
In the temperature-dependent emission spectra, an increase in the temperature from 10 ℃ to 40 ℃ in dichloromethane led to a decrease in the intensity of the emission (Fig. S3b), which was due to theincreaseof thenonradioactive decayas a resultof the aggravation of the movement of the rotaxane. Meanwhile, upon increasing the concentration from 1.0 × 10-6 mol/L to 1.0 × 10-4 mol/L in dichloromethane, the peak was red shifted (Fig. 3b), giving a further improvement of J-type aggregation of the zinc porphyrin core.
With the aim to obtaining more information on the aggregation of [5]rotaxane in solvents, the concentration-dependent 1H NMR spectra of the [5]rotaxane were recorded. In the concentration-dependent 1H NMR experiment in dichlormethane-d2 (Fig. S2 in Supporting information), the increase of concentration resulted in the slight downfield shifts of the resonance signals for the aromatic protons on the zinc porphorin core and the adjacent benzyl rings. The concentration-dependent 1H NMR experiment results confirmed that the aggregation of the zinc porphyrin core in dichloromethane was driven by π–π interactions. The aggregation of the [5]rotaxane was further examined by both scanning electron microscopy (SEM) and transmission electron microscope (TEM). Interestingly, the [5]rotaxane exhibited large-scale spheres upon being exposed to the non-polar solvent (DCM:n-hexane = 1: 9, v/v) followed by slow evaporation in air at room temperature (Fig. 4a and Fig. S4 in Supporting information). Moreover, the sized distribution as shown in Fig. 4a was from 0.22 mm to 0.41 mm, with an averaged size of 0.32 mm. The TEM images of the spheres indicated that the spheres were solid rather than hollow. However, while the used solvent was changed to the polar solvent (DCM: acetone = 1:9, v/v), the morphology of the [5]rotaxane changed into the uniform and highly ordered fibrous network structure (Fig. 4c and Fig. S5 in Supporting information). The width of the nanofiber was ca. 80 nm. The different morphologies indicated that the polarity of the solvents played an important role in the formation of selfassembled nanostructures.

|
Download:
|
| Fig. 4. SEM images of compound 1 prepared in a mixture of (a) DCM and n-hexane (1:9, v/v) and (c) DCM and acetone (1:9, v/v); compound 2 prepared in a mixture of (b) DCM and n-hexane (1:9, v/v) and (d) DCM and acetone (1:9, v/v). The inset image in (a) showed TEM results of compound 1 prepared in a mixture of dichloromethane and n-hexane (1:9, v/v). SEM images of compound 1 prepared in a mixture of DCM and acetone (1:9, v/v) after aging for 0 h (e), 12 days (f), 16 days (g) and 40 days (h). Scale bars: 2.0 μm. | |
{kind=link}
Surprisingly, the model compound 2, which featured the almost same structure without DPP[5]A, showed the irregular morphology in the same solvents (Figs. 4b, d, and Figs. S6 and S7 in Supporting information). It is speculated that the existence of the DPP[5]A limited the movement of the long alkane chains, that is, to some extent, reduced the disorder of the soft chains, and thus promoted the aggregation, and finally formed the regular morphology. This phenomenon indicated that the interlocked structure was indispensable for the formation of the ordered nanostructures. The pillarenes made the alkyl chains rigid and thus led the ordered aggregation of whole [5]rotaxane.
Moreover, it is interesting that the compound 1 exhibited a morphology evolution in a mixture of DCM and acetone (1:9, v/v). The morphology evolved from nanofibers to nanospheres after half month (Figs. 4e–h and Figs. S8–S22 in Supporting information). From the morphology recorded by time, it could be deduced that the nanospheres were formed from tangle of nanofibers. To exclude the influence of solvent polarity, a series of solution of compound 1 in different ratio of dichloromethane and acetone was prepared. The ratio of dichloromethane and acetone varied from 1:1 to 1:99 (Figs. S23–S32 in Supporting information). The results of the solvent-ratio-dependent morphology excluded the effect of polarity changes as time went on.
From the microscopy images and all other spectroscopic changes observed, the possible mechanisms for the aggregation process in different solvents were suggested in Fig. S33 in Supporting information. Firstly, the complex was dissolved in dichloromethane and formed the oligomer with the help of π–π stacking. Then, the polarity of solvent played an important role in the forming of the morphology. In non-polar solution, the oligomers tended to pack close and form spheres with the minimal superficial area. When being deposed into acetone with a relatively large polarity, the oligomers tended to relatively stretch and thus generated the nanofibers. Meanwhile, as time went on, the nanofibers enwinded with each other and led to the morphology evolution into nanoshperes.
In conclusion, we reported the successful synthesis of a highly ordered and D4h symmetric [5]rotaxane with zinc porphyrin as the core, platinum-acetylide as linkage, and four arms decorated with pillararene from a convenient method. The target [5]rotaxane was well characterized with NMR spectroscopy, mass spectrometer, and PM6 semiempirical molecular orbital methods. Meanwhile, the intramolecular energy transfer from platinum-acetylide to the zinc porphyrin was observed. The detailed investigation on UV–vis spectrum and emission spectrum indicated the formation of a J-type aggregation in dichloromethane driven by π–π interactions. The [5]rotaxane formed an ordered solid nanospheres with an average diameter of ca. 320 nm in non-polar solvent but nanofibers in polar solvent, while the similar structure without pillar[5]arene aggregated amorphously under the same condition due to the reduce of disorder of the soft alkane chains by pillar[5]arene. Furthermore, the nanofibers were obtained in polar solvent, which thus evolved into nanoshperes after half month along with the tangle of the nanofibers.
AcknowledgmentsH.-B. Yang acknowledges the financial support from the National Natural Science Foundation of China (No. 21572066), STCSM (No. 16XD1401000), and Program for Changjiang Scholars and Innovative Research Team in University.
Appendix A. Supplementary dataSupplementarymaterial related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.10.035.
| [1] |
J.P. Sauvage, Acc. Chem. Res. 31 (1998) 611-619. DOI:10.1021/ar960263r |
| [2] |
X. Han, G. Liu, S.H. Liu, J. Yin, Org. Biomol. Chem. 14 (2016) 10331-10351. DOI:10.1039/C6OB01581F |
| [3] |
M. Fujita, Acc. Chem. Res. 32 (1999) 53-61. DOI:10.1021/ar9701068 |
| [4] |
R.J. Puddephatt, J. Organomet. Chem. 792 (2015) 13-24. DOI:10.1016/j.jorganchem.2014.12.003 |
| [5] |
Y. Liu, M. O'Keeffe, M.M.J. Treacy, O.M. Yaghi, Chem. Soc. Rev. 47 (2018) 4642-4664. DOI:10.1039/C7CS00695K |
| [6] |
S.A. Nepogodiev, J.F. Stoddart, Chem. Rev. 98 (1998) 1959-1976. DOI:10.1021/cr970049w |
| [7] |
L. Chen, Q. Chen, M. Wu, F. Jiang, M. Hong, Acc. Chem. Res. 48 (2015) 201-210. DOI:10.1021/ar5003076 |
| [8] |
N.H. Evans, Chem.-Eur. J. 24 (2017) 3101-3112. |
| [9] |
G. Gil-Ramírez, D.A. Leigh, A.J. Stephens, Angew. Chem. Int. Ed. 54 (2015) 6110-6150. DOI:10.1002/anie.201411619 |
| [10] |
M. Xue, Y. Yang, X. Chi, X. Yan, F. Huang, Chem. Rev. 115 (2015) 7398-7501. DOI:10.1021/cr5005869 |
| [11] |
J.E.M. Lewis, M. Galli, S.M. Goldup, Chem. Commun. 53 (2017) 298-312. DOI:10.1039/C6CC07377H |
| [12] |
M. Marenda, E. Orlandini, C. Micheletti, Nat. Commun. 9 (2018) 3051.
|
| [13] |
H.N. Zhang, W.X. Gao, Y.X. Deng, Y.J. Lin, G.X. Jin, Chem. Commun. 54 (2018) 1559-1562. DOI:10.1039/C7CC09448E |
| [14] |
S.L. Huang, Y.J. Lin, Z.H. Li, G.X. Jin, Angew. Chem. Int. Ed. 53 (2014) 11218-11222. DOI:10.1002/anie.201406193 |
| [15] |
T. Kim, N. Singh, J. Oh, et al., J. Am. Chem. Soc. 138 (2016) 8368-8371. DOI:10.1021/jacs.6b04545 |
| [16] |
Y. Lu, H.N. Zhang, G.X. Jin, Acc. Chem. Res. 51 (2018) 2148-2158. DOI:10.1021/acs.accounts.8b00220 |
| [17] |
H. Wang, Z.J. Zhang, H.Y. Zhang, Y. Liu, Chin. Chem. Lett. 24 (2013) 563-567. DOI:10.1016/j.cclet.2013.04.007 |
| [18] |
G. Yu, D. Wu, Y. Li, et al., Chem. Sci. 7 (2016) 3017-3024. DOI:10.1039/C6SC00036C |
| [19] |
J.F. Chen, B.B. Han, J.F. Ma, et al., RSC Adv. 7 (2017) 47709-47714. DOI:10.1039/C7RA10326C |
| [20] |
A. Brown, T. Lang, K.M. Mullen, P.D. Beer, Org. Biomol. Chem. 15 (2017) 4587-4594. DOI:10.1039/C7OB01040K |
| [21] |
R. Tepper, U.S. Schubert, Angew. Chem. Int. Ed. 57 (2018) 6004-6016. DOI:10.1002/anie.201707986 |
| [22] |
M.J. Langton, P.D. Beer, Acc. Chem. Res. 47 (2014) 1935-1949. DOI:10.1021/ar500012a |
| [23] |
P.A. Gale, E.N.W. Howe, X. Wu, Chem. Commun. 47 (2011) 82-86. DOI:10.1039/C0CC00656D |
| [24] |
M. Denis, J. Pancholi, K. Jobe, M. Watkinson, S.M. Goldup, Angew. Chem. Int. Ed. 57 (2018) 5310-5314. DOI:10.1002/anie.v57.19 |
| [25] |
Y.J. Lee, K.S. Liu, C.C. Lai, et al., Chem.-Eur. J. 23 (2017) 9756-9760. DOI:10.1002/chem.201702525 |
| [26] |
K. Eichstaedt, J. Jaramillo-Garcia, D.A. Leigh, et al., J. Am. Chem. Soc. 139 (2017) 9376-9381. DOI:10.1021/jacs.7b04955 |
| [27] |
G. De Bo, M.A.Y. Gall, S. Kuschel, et al., Nat. Nanotechnol. 13 (2018) 381-385. DOI:10.1038/s41565-018-0105-3 |
| [28] |
M. Centola, J. Valero, M. Famulok, J. Am. Chem. Soc. 139 (2017) 16044-16047. DOI:10.1021/jacs.7b08839 |
| [29] |
S. Dong, J. Yuan, F. Huang, Chem. Sci. 5 (2014) 247-252.
|
| [30] |
H. He, E.M. Sevick, D.R.M. Williams, J. Chem. Phys. 148 (2018) 134905.
|
| [31] |
A. Goujon, T. Lang, G. Mariani, et al., J. Am. Chem. Soc. 139 (2017) 14825-14828. DOI:10.1021/jacs.7b06710 |
| [32] |
Y.W. Yang, Y.L. Sun, N. Song, Acc. Chem. Res. 47 (2014) 1950-1960. DOI:10.1021/ar500022f |
| [33] |
D. Wendinger, R.R. Tykwinski, Acc. Chem. Res. 50 (2017) 1468-1479. DOI:10.1021/acs.accounts.7b00164 |
| [34] |
J.K. Szymanski, Y.M. Abul-Haija, L. Cronin, Acc. Chem. Res. 51 (2018) 649-658. DOI:10.1021/acs.accounts.7b00495 |
| [35] |
Á. Martínez, C. Ortiz Mellet, J.M. García Fernández, Chem. Soc. Rev. 42 (2013) 4746-4773. DOI:10.1039/c2cs35424a |
| [36] |
S. Xiong, X. Zhang, L.B. Meng, et al., Chem. Commun. 51 (2015) 6504-6507. DOI:10.1039/C5CC01345C |
| [37] |
C. Gao, Z.L. Luan, Q. Zhang, et al., Org. Lett. 19 (2017) 1618-1621. DOI:10.1021/acs.orglett.7b00393 |
| [38] |
Q.S. Fang, L. Chen, Q.Y. Liu, Chin. Chem. Lett. 28 (2017) 1013-1017. DOI:10.1016/j.cclet.2016.12.002 |
| [39] |
S.J. Zhang, Q. Wang, M. Cheng, et al., Chin. Chem. Lett. 26 (2015) 885-888. DOI:10.1016/j.cclet.2015.05.015 |
| [40] |
L.L. Hu, W. Xue, J. Yin, Chin. Chem. Lett. 27 (2016) 155-158. DOI:10.1016/j.cclet.2015.09.010 |
| [41] |
Z. Meng, Y. Han, L.N. Wang, et al., J. Am. Chem. Soc. 137 (2015) 9739-9745. DOI:10.1021/jacs.5b05758 |
| [42] |
X.Q. Wang, W. Wang, W.J. Li, et al., Nat. Commun. 9 (2018) 3190.
|
| [43] |
W. Wang, L.J. Chen, X.Q. Wang, et al., Proc. Natl. Acad. Sci. U. S. A. 112 (2015) 5597-5601. DOI:10.1073/pnas.1500489112 |
| [44] |
Z.J. Zhang, H.Y. Zhang, H. Wang, Y. Liu, Angew. Chem. Int. Ed. 50 (2011) 10834-10838. DOI:10.1002/anie.v50.46 |
| [45] |
Z. Meng, J.F. Xiang, C.F. Chen, Chem. Sci. 5 (2014) 1520-1525. DOI:10.1039/c3sc53295j |
| [46] |
S.J. Rao, Q. Zhang, X.H. Ye, C. Gao, D.H. Qu, Chem.-Asian J. 13 (2018) 815-821. DOI:10.1002/asia.201800011 |
| [47] |
M.H. Ding, X.M. Chen, L.L. Tang, F. Zeng, Chin. Chem. Lett. 28 (2017) 1375-1379. DOI:10.1016/j.cclet.2017.03.009 |
| [48] |
W. Wang, Y. Zhang, B. Sun, et al., Chem. Sci. 5 (2014) 4554-4560. DOI:10.1039/C4SC01550A |
| [49] |
W. Wang, B. Sun, X.Q.Q. Wang, et al., Chem.-Eur. J. 21 (2015) 6286-6294. DOI:10.1002/chem.v21.16 |
| [50] |
Y.X. Wang, Q.F. Zhou, L.J. Chen, et al., Chem. Commun. 54 (2018) 2224-2227. DOI:10.1039/C7CC08729B |
| [51] |
X.D. Xu, J. Zhang, L.J. Chen, et al., Chem. Commun. 48 (2012) 11223-11225. DOI:10.1039/c2cc36371b |
| [52] |
G. Ji, Z. Yang, Y. Zhao, et al., Chem. Commun. 51 (2015) 7352-7355. DOI:10.1039/C5CC00609K |
| [53] |
K. Onitsuka, H. Kitajima, M. Fujimoto, et al., Chem. Commun. (2002) 2576-2577.
|