 2019, Vol. 30
2019, Vol. 30 


In the past decades, supramolecular polymers have been an attractive topic in supramolecular chemistry due to their dynamic nature and high responsiveness to outside stimuli [1]. Currently, hydrogen bonding [2], metal coordination [3], donor-acceptor interaction [4], and hydrophobicity [5] have been extensively used for driving the generation of supramolecular polymers. Nevertheless, in most cases, supramolecular polymers can exist only in organic solvents. Further application of supramolecular polymers as biomaterials would require high stability and solubility in water to enable good biocompatibility [5, 6]. In this context, hydropho-bicity-driven encapsulation of cyclodextrins and cucurbiturils for hydrophobic monomeric or dimeric species has been used to produce various cross-linked supramolecular polymers in aqueous solution [5]. Another more straightforward approach involves the use of ion-pair electrostatic attraction, which can provide both good water-solubility and biocompatibility. However, single ionpair interaction is not able to allow for efficient binding due to the competitiveness of water. Schmuck et al. has successfully combined ion-pair electrostatic attraction and multiple hydrogen bonds to generate switchable supramolecular systems in polar solvents [7]. Previously, we have developed a novel motif of mutually promoting electrostatic (ion-pair) attraction and aromatic stacking in water [8], which has been used to assemble supramolecular polymers in water from tetrapyridinium and diskshaped coronene multicarboxylate monomers [9]. Because it has been well-established that multitopic building blocks exhibit enhanced binding through multivalency [10], we become interested in the self-assembly of two discrete multitopic monomers into supramolecular networks through this cooperative binding motif. We herein report the generation of a new kind of supramolecular polymers in water from tetrapyridinium TP and tetraphenylethylene derivatives TPE-1~3 (Fig. 1).

|
Download:
|
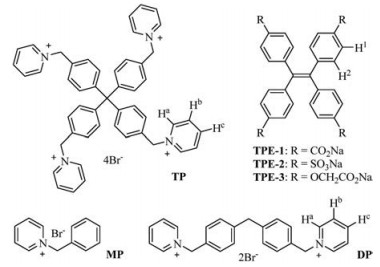
| Fig. 1. The structures of ionic compounds TPE-1~3, TP, MP and DP. | |
Highly water-soluble tetraphenylethylene derivatives TPE-1~3 were chosen as aromatic multianionic monomers for their studying electrostatic and stacking interactions with water-soluble tetracationic monomer TP [10] in water. For comparison, dipyridinium DP [11] was also prepared as bromide salt. The synthesis and characterization details for TPE-1~3 are provided in the Supporting information.
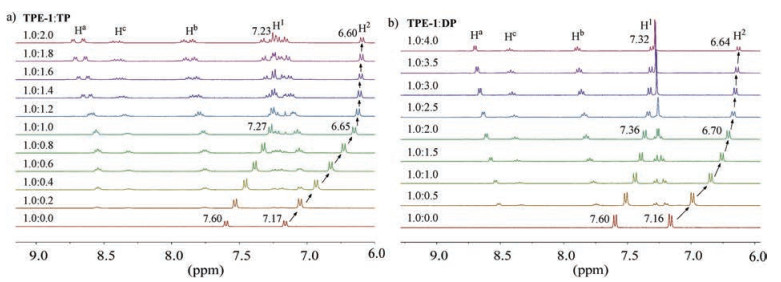
1H NMR titration experiments were first performed by adding increasing amounts of TP or DP to the solution of TPE-1~3 in D2O. Adding TP (0–2.0 mmol/L) to the solution of TPE-1 (1.0 mmol/L) in D2O caused dramatic upfield shifting of the H-1 and H-2 signals of TPE-1 (Fig. 2a). Notably, the addition of 1.0 equiv. of TP could cause the maximum shifting, about –0.33 ppm for H-1 and –0.52 ppm for H-2, respectively. Further increase of TP to 2.0 equiv. only led to small shifting (< –0.05 ppm). This result supported that the binding between the two compounds occurred in a 1:1 stoichiometry. Similar shifting tendency was also observed when adding TP to the solution of TPE-2 or TPE-3 in D2O. For TPE-2, 1.0 equiv. of TP caused its H-1 and H-2 signals to shift upfield by –0.37 and –0.54 ppm, respectively, and for TPE-3, the values were –0.45 and –0.49 ppm, respectively (Figs. S1 and S2 in Supporting information). 1H NMR titration experiments were also conducted for TPE-1 with the addition of DP in D2O (Fig. 2b). It can be found that 2.0 equivalent of DP could cause maximum upfield shifting (–0.24 and –0.45 ppm, respectively) for the H-1 and H-2 signals of TPE-1, and further addition of another 2.0 equiv. led to very small shifting (< 0.06 ppm), supporting their binding was in a 1:2 stoichiometry. As expected, 1H NMR experiments for the mixture of DP with TPE-2 or TPE-3 gave rise to similar results (Figs. S3 and S4 in Supporting inforamtion). In contrast, adding 8.0 equiv. of MP to the solution of TPE-1 or TPE-3 (1.0 mmol/L) in D2O caused only up to –0.09 ppm of upfield shifting for their H-1 and H-2 signals in the 1H NMR spectra (Fig. S5 in Supporting information), indicative of much weaker binding.

|
Download:
|
| Fig. 2. Partial 1H NMR (400 MHz) spectra of the mixtures of a) TPE-1 (1.0 mmol/L) and TP (0–2 mmol/L) and b) TPE-1 (1.0 mmol/L) and DP (0–4 mmol/L) in D2O at 25 ℃. | |
Fluorescence emission experiments were then performed for the three mixtures of TP and TPE-1~3. Significant fluorescence quenching was found for all three tetraphenylethylene derivatives by TP (Fig. S6 in Supporting information). Again, 1.0 equiv. of TP could lead to the maximum quenching, which was consistent with the above 1H NMR experiments and again supported the 1:1 stoichiometry. Given the low concentration of both samples, this observation reasonably evidenced high binding strength between the two multitopic samples. Fluorescence titration experiments for TPE-1~3 by DP were also performed (Fig. S6), which revealed continuous quenching of the fluorescence of TPE-1. However, no quenching saturation was observed after 4.0 equiv. of DP was added, reflecting relatively lowered binding strength. Adding 8.0 equiv. of MP to the solution of TPE-1 or TPE-3 caused nearly no quenching for their fluorescence, indicating that at low concentration, no binding occurred.
Concentration-varying fluorescence experiments showed that the three tetraphenylethylene tetraanion did not exhibit aggregation-induced emission [12], probably due to the electrostatic repulsion of the anions. UV–vis absorption spectra were further recorded for the mixtures of TPE-1 and TP as well as DP by keeping a constant total concentration (Fig. S7 in Supporting informaiton). Job's plot was thus obtained for the first mixture by recording the absorbance change of the TPE unit at 295 nm (Fig. S7), which further supported the 1:1 binding stoichiometry. Job's plot could not be obtained from similar UV–vis experiments for the mixture of TPE-1 and DP because of limited absorbance change of the TPE unit. We thus adopted the fluorescence approach and Job's plot obtained supported the 1:2 binding stoichiometry (Fig. S8 in Supporting information). As expected, Job's plots obtained using similar absorption and fluorescence methods supported 1:1 stoichiometry for the mixture of TP with TPE-2 or TPE-3 and 1:2 stoichiometry for the mixture of DP with either of them (Figs. S9-S14 in Supporting inforamtion). Because TPE-1~3 and TP have four ionic aromatic units, whereas DP has two, the above binding stoichiometry clearly pointed to a one-to-one ion-pair binding motif.
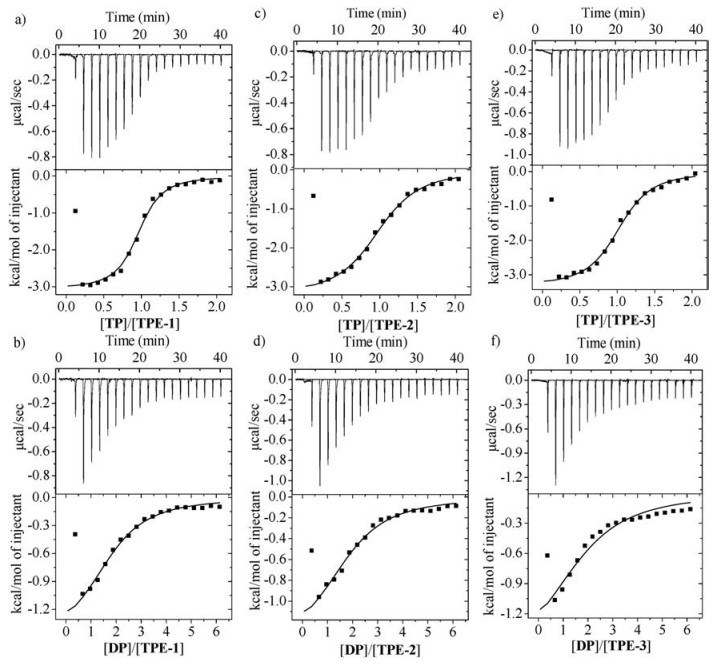
Isothermal titration calorimetric (ITC) experiments were then carried out to quantitatively evaluate the binding behavior of TPE-1~3 with TP or DP (Fig. 3). The data of the apparent association constants (Ka), Gibbs energy changes (△G) and associated enthalpy and entropy changes are summarized in Table 1. Because the above fluorescence experiments revealed that at low concentrations, no important binding occurred for monotopic MP, the quite high association constants exhibited by ditopic DP supported that the two pyridinium units of DP cooperated in binding the TPE derivatives, reasonably through folding into a cleft conformation. This folding conformation might interact with one or two appended benzene rings of the TPE monomers, even though currently we could differentiate them. Such a binding motif suggested that the mixtures of DP with the TPE derivatives did not produce any kind of supramolecular polymers. By assuming that the two cationic molecules adopted the identical binding motif, we might expect that tetratopic TP would form linear supramolecular polymers with the tetraanions. For all the three tetraanions, TP displayed considerably higher binding affinity than DP, which showed that the supramolecular polymers formed by TP might further undergo aggregation to allow for binding enhancement. For all the six mixtures, the contribution of -T△S was larger than DH, which reflected that the release of the "confined" water molecules exposed to the hydrophobic surface of the aromatic units as a result of stacking produced the largest driving force for the binding of the opposite ions [8, 13]. For the mixtures of DP and TP with the identical TPE derivative, the -T△S values were comparable. However, the DH of the mixture of TP was all pronouncedly larger than that of DP, which we tentatively attributed to the enhanced electrostatic attraction due to the further aggregation of the supramolecular polymers formed by TP.

|
Download:
|
| Fig. 3. ITC measurements for the titration of TP (1 mmol/L in water) into water solution of TPE-1 (0.1 mmol/L) (a), TPE-2 (0.1 mmol/L) (c), TPE-3 (0.1 mmol/L) (e). DP (3 mmol/L in water) into water solution of TPE-1 (0.1 mmol/L) (b), TPE-2 (0.1 mmol/L) (d), TPE-3 (0.1 mmol/L) (e). | |
|
|
Table 1 Summary of the apparent association constants and related thermodynamic data in water. |

{kind=link}
{kind=link}
{kind=link}
Finally, dynamic light scattering (DLS) experiments were conducted in water for all the above six mixtures, with the molar ratio being controlled to be consistent with the binding stoichiometry. The three mixtures of TPE/DP (1:2, [DP] = 1.0 mmol/L) gave rise to an average hydrodynamic diameter (DH) of 1.5, 1.6 and 2.0 nm (Fig. S15 in Supporting inforamtion), respectively, which was consistent with the formation of simple 1:2 complexes. In contrast, the three mixtures of TPE/TP (1:1, [TP] = 0.5 mmol/L) afforded much larger DH of 255, 295 and 342 nm, respectively. The values clearly supported the formation of large supramolecular entities. As expected, with the increase of the concentration, the DH values increased pronouncedly (Fig. S16 in Supporting information). The above fluorescence and ITC experiments supported that these three mixtures formed linear supramolecular polymers, even though they should be randomarranged and other forms of supramolecular entities could not be excluded. Because the apparent association constant of these three systems was all considerably higher than that of the corresponding complex of DP, we propose that the linear supramolecular polymers underwent further aggregation to produce 3D networks, even though the refined binding motif cannot be established due to the dynamic and low directionality of electrostatic and stacking interactions.
In conclusion, we have constructed a new family of supramolecular polymers in water from tetrahedral tetracationic and tetraanionic monomers and demonstrated the efficiency of the combination of electrostatic and aromatic stacking interactions in stabilizing the supramolecular polymers. The important contribution of the stacking interaction raises the possibility of utilizing the stacking between neutral and hydrophobic electron-rich and deficient aromatic units for the generation of water-soluble more ordered one-dimensional supramolecular polymers, from which controlled cross-layer-relayed electron transfer is expected.
AcknowledgmentWe thank the National Natural Science Foundation of China (Nos. 21432004 and 21472023) for financial support.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.10.016.
| [1] |
(a) J.M.Lehn, Polym.Int.51 (2002)825-839 (b) L.Brunsveld, B.J.B.Folmer, E.W.Meijer, R.P.Sijbesma, Chem.Rev.101 (2001)4071-4097 (c) X.Zheng, Q.Miao, W.Wang, D.H.Qu, Chin.Chem.Lett.29 (2018)1621-1624 (d) X.Wang, Y.Yang, L.Fan, F.Yang, D.Wu, Sci.China Chem.61 (2018)311-318 (e) G.Yin, L.Chen, C.Wang, H.Yang, Chin.J.Chem.36 (2018)134-138 (f) C.Xu, L.Xu, X.Ma, Chin.Chem.Lett.29 (2018)970-972 (g) Q.Zhao, Y.Chen, Y.Liu, Chin.Chem.Lett.29 (2018)84-86 (h) C.Xiong, R.Sun, Chin.J.Chem.35 (2017)1669-1672 (i) Z.J.Yin, Z.Q.Wu, F.Lin, et al., Chin.Chem.Lett.28 (2017)1167-1171 (j) Q.Wang, M.Cheng, J.L.Jiang, L.Y.Wang, Chin.Chem.Lett.28 (2017)793-797 (k) P.Wang, J.Ma, D.Xia, Org.Chem.Front.5 (2018)1297-1302 (l) M.Y.Li, K.Han, J.Li, X.S.Jia, C.J.Li, Acta Polym.Sin. (2017)129-134 (m) C.W.Zhang, B.Ou, G.Q.Yin, L.J.Chen, H.B.Yang, Acta Polym.Sin. (2017)71-79 (n) Z.S.Yang, Y.K.Tian, Z.J.Li, et al., Acta Polym.Sin. (2017)121-128 (o) Y.Fan, F.Lin, X.N.Xu, J.Q.Xu, X.Zhao, Acta Polym.Sin. (2017)80-85 (p) M.Liu, G.Ouyang, D.Niu, Y.Sang, Org.Chem.Front.5 (2018)2885-2900 (q) X.F.Ji, D.Y.Xia, X.Z.Yan, H.Wang, F.H.Huang, Acta Polym.Sin. (2017)9-18 (r) L.Yang, X.Tan, Z.Wang, X.Zhang, Chem.Rev.115 (2015)7196-7239. |
| [2] |
(a) S.Chen, W.H.Binder, Acc.Chem.Res.49 (2016)1409-1420 (b) B.Isare, S.Pensec, M.Raynal, L.Bouteiller, Compt.Rendus Chim.19 (2016)148-156 (c) M.Anthamatten, Adv.Polym.Sci.268 (2015)47-99. |
| [3] |
(a) A.Winter, U.S.Schubert, Chem.Soc.Rev.45 (2016)5311-5357 (b) H.Li, L.Wu, Soft Matter 10 (2014)9038-9053. |
| [4] |
(a) W.Hayes, B.W.Greenland, Adv.Polym.Sci.268 (2015)143-166 (b) Y.K.Tian, Y.F.Han, Z.S.Yang, F.Wang, Macromolecules 49 (2016)6455-6461 (c) B.Zheng, F.Wang, S.Dong, F.Huang, Chem.Soc.Rev.41 (2012)1621-1636. |
| [5] |
(a) A.Harada, Y.Takashima, M.Nakahata, Acc.Chem.Res.47 (2014)2128-2140 (b) J.Tian, L.Zhang, H.Wang, D.W.Zhang, Z.T.Li, Supramol.Chem.28 (2016)769-783 (c) Y.Liu, H.Yang, Z.Wang, X.Zhang, Chem.Asian J.8 (2013)1626-1632. |
| [6] |
(a) X.Ma, H.Tian, Acc.Chem.Res.47 (2014)1971-1981 (b) Z.Ding, H.Li, W.Gao, et al., Chin.J.Chem.35 (2017)447-456 (c) E.Krieg, M.M.C.Bastings, P.Besenius, B.Rybtchinski, Chem.Rev.116 (2016)2414-2477 (d) Q.Zhao, Y.Chen, Y.Liu, Chin.Chem.Lett.29 (2018)84-86. |
| [7] |
(a) C.Schmuck, W.Wienand, J.Am.Chem.Soc.125 (2003)452-459 (b) C.Schmuck, K.Samanta, M.Ehlers, Chem.Eur.J.22 (2016)15242-15247. |
| [8] |
(a) Y.C.Zhang, Y.Qin, H.Wang, et al., Chem.Asian J.11 (2016)1065-1070 (b) Y.Zhang, S.B.Yu, B.Yang, et al., Org.Chem.Front.5 (2018)1039-1044. |
| [9] |
Q. Qi, B. Yang, H. Wang, D.W. Zhang, Z.T. Li, Tetrahedron 74 (2018) 2792-2796. DOI:10.1016/j.tet.2018.04.047 |
| [10] |
(a) A.Mulder, J.Huskens, D.N.Reinhoudt, Org.Biomol.Chem.2 (2004)3409-3424 (b) C.Fasting, C.A.Schalley, M.Weber, et al., Angew.Chem.Int.Ed.51 (2012)10472-10498 (c) C.Yao, J.Tian, H.Wang, et al., Chin.Chem.Lett.28 (2017)893-899 (d) H.Wang, D.W.Zhang, Z.T.Li, Acta Polym.Sin. (2017)19-26 (e) J.Tian, C.Yao, W.L.Yang, et al., Chin.Chem.Lett.28 (2017)798-806 (f) X.F.Li, S.B.Yu, B.Yang, et al., Sci.China Chem.61 (2018)830-835 (g) Y.Chen, F.Huang, Z.T.Li, Y.Liu, Sci.China Chem.61 (2018)979-992 (h) T.K.Dam, M.L.Talaga, N.Fan, C.F.Brewer, Methods Enzym.567 (2016)71-95 (i) J.Yan, W.Li, A.Zhang, Chem.Commun.50 (2014)12221-12233; (j) E.Lopez-Fontal, L.Milanesi, S.Tomas, Chem.Sci.7 (2016)4468-4475. |
| [11] |
J.M. Campos, M.C. Nunez, R.M. Sanchez, et al., Bioorg.Med.Chem. 10 (2002) 2215-2231. |
| [12] |
(a) Y.Hong, J.W.Y.Lam, B.Z.Tang, Chem.Soc.Rev.40 (2011)5361-5388 (b) Y.Shi, Y.Cai, Y.J.Wang, et al., Sci.China Chem.60 (2017)635-641 (c) S.Liu, J.Cheng, J.Xu, Chin.J.Chem.35 (2017)335-340 (d) X.Y.Teng, X.C.Wu, Y.Q.Cao, et al., Chin.Chem.Lett.28 (2017)1485-1491 (e) Y.Song, L.Zong, L.Zhang, Z.Li, Sci.China Chem.60 (2017)1596-1601. |
| [13] |
T.P.Silverstein, J.Chem.Educ.75 (1998)116-118.
|