 2019, Vol. 30
2019, Vol. 30 


b China MS Center, Shimadzu(China) Co., Beijing Branch, Beijing 100020, China
Sample preparation plays a very important role in the total chemical analytical process. It takes a lot of time and labor to separate trace amount of analyte from the complex matrices. Besides the conventional solvent extraction method, many off-line or on-line preparation techniques have been developed to reduce the total analytical time or increase the extraction efficiency [1-4]. Especially, supercritical fluid extraction (SFE) has been emerging as a strong extraction technique [5-8]. Use of supercritical fluid make it possible to reduce extraction time and organic solvents consumption. Among the supercritical fluids, supercritical CO2 (SC-CO2) is the most widely used for the SFE technique because of its low critical temperature (31 ℃) and pressure (72 bar), chemically inertness, low toxicity and non-flammability. In addition, it is cheap and readily available in bulk quantities with a high degree of purity.
As an alternative to traditional extraction methods, SC-CO2 extraction technology has been widely applied in various fields, such as food science [9-11], natural products [12-14], pharmaceutical [15, 16] and environmental science [17, 18]. It was also coupled to analytical or preparative supercritical fluid chromatography (SFC) for on-line extraction and separation of fat-soluble vitamins [19, 20], natural aromatic substances [21], traditional Chinese medicine [22, 23], disease biomarkers [24] and environmental pollutants [25, 26]. Moreover, recently, SFE [27-31] and SFC [32] were also coupled to the reverse phase liquid chromatography (RPLC), respectively, since RPLC can provide different separation selectivity for better resolution. However, only a few applications on SFE-RPLC have been reported, since coupling SFE-RPLC is technically difficult and the decompressed CO2 gas introduced into the LC system could make the flow rate unstable and the peak shape bad. In this study, on-line SFE was coupled to the RPLC/MS by using a trap column and a high pressure six-port valve. A dilution line was used to on-line concentrate the extract and improve the peak shape. Furthermore, this system was successfully applied to on-line extraction and reverse phase separation for capsaicinoids in several kinds of capsicum fruits.
In this study, capsaicin was purchased from The National RM Resources Network (Beijing, China). Methanol was purchased from Fisher Scientific (Glee, Belgium). All of the solvents were of LC–MS grade. Pure water was prepared from Milli-Q Advantage A10 system (Merck Millipore, MA, USA). The extraction dispersant, Miyazaki hydro protect, was provided by Shimadzu (Tokyo, Japan).
All experiments were performed on a Nexera UC system(Shimadzu, Japan), consisting of a CBM-20 A control bus module, an SFE-30 A autoextractor with a rack changer, a DGU-20 Adegasser, an LC-30ADSF (SC-CO2 delivery pump), four LC-20ADXR (liquid delivery pumps for modifier,diluting solventandmobilephase),aCTO-20ACcolumnoven, ahighpressuresix-portvalve,aSFC-30 Abackpressure regulator(BPR). An SPD-M20 A photo diode array ultraviolet spectroscopic detector is attached when it is necessary. LCMS-8050 Triple Quadrupole mass spectrometer (Shimadzu, Japan) is attached. All data was processed by Shimadzu Labsolution Ver. 5.8.6 software (Shimadzu, Japan).
Shim-pack VP-ODS (4.6 mm I.D. × 50 mm, particle size 5 mm, Shimadzu, Japan) was selected as a trap column. Cartridge Guard Column C18 (4.0 mm I.D. ×10 mm, particle size 5 mm, GL Sciences Inc., Japan) was selected as a concentration column. The analysis column was Shim-pack GIST-HP C18 (3.0 mm I.D. ×100 mm, particle size 3 mm, Shimadzu, Japan).
The schematic diagram of on-line SFE-LC/MS system is shown in Fig. 1. The high pressure six port valve is centered in this system and left hand is SFE part and right hand is LC/MS part.

|
Download:
|
| Fig. 1. Schematic diagram of on-line SFE-LC/MS system. (a) The high pressure valve was set at position (a); (b) The high pressure valve was set at position (b). 1: CO2 cylinder, 2: modifier, 3: CO2 pump, 4: liquid delivery pump, 5: SFE unit, 6: SFE vessel, 7: BPR, 8: trap column, 9: dilute solvent, 10: high pressure six port valve, 11: drain, 12: concentration column, 13: analytical column, 14: mobile phase, 15: detector (MS/PDA). | |
{kind=link}
First, the high pressure valve was set at position (a), as shown in Fig. 1a. In SFE part, BPR pressurewas set to be 15 MPa. The fat-soluble components were extracted by SC-CO2 with 5% methanol as the modifier, by using a closed extraction vessel (static extraction, 0– 4 min). Then, the supercritical fluid carried the extracts from the vessel through the BPR (dynamic extraction, 4–8 min) into the trap column at a total flow rate of 5 mL/min. After completing the entire extractionprocess, CO2 pumpwas stopped andflow rate ofmethanol was changed from 0.25 mL/min to 0.5mL/min to elute the extracts quickly. Flow rate of the dilution line (100% water) was set to be 1.5mL/min and the extracts were concentrated on the concentration column. After 10 min, the valve was changed from position (a) to position (b) (Fig. 1b). The concentrated extractswere flushedinto the separation column. Separation was performed by using a binary gradient method. Mobile phase consisted of water (A) and methanol (B), and the gradient condition was as follows: 0 min (B) 45% - > 10 min (B) 80% - > 12 min (B) 100% - > 13.5 min (B) 100% - > 13.6 min (B) 45%. Flow rate is 0.4 mL/min. The column temperature is 50 ℃. Data acquisition was done in positive ion electrospray ionization (ESI) mode under the following conditions: capillary voltage: 4 kV; interface temperature: 300 ℃; DL temperature: 250 ℃; heat block temperature: 450 ℃; nebulizing gas flow: 3.0 L/min; heating gas flow: 10 L/min; drying gas flow: 10 L/min. Multiple reaction monitoring (MRM) was employed in this study.
100 mg of green, yellow and red bell pepper samples were well mixed with the Nexera UC extraction dispersant, and then filled into three 5-mL extraction vessels, respectively. Purpose of mixing with the dispersant was to improve sample-solvent contact, and to avoid channeling when CO2 flowed through the mixture.
Typical capsaicinoids are constructed by the vanillyl base from vanillin. Then all of them have 137 m/z product ion. Multiple reaction monitoring (MRM) was employed in this study (capsaicin 306.4 > 137, 306.4 > 122, 306.4 > 170, 306.4 > 182; dihydrocapsaicin 308.4 > 137, 308.4 > 122, 308.4 > 184; nordihydrocapsaicin 294.4 > 137, 294.4 > 122, 294.4 > 170; homocapsaicin 320.4 > 137, 320.4 > 122, 320.4 > 196; homodihydrocapsaicin 322.5 > 137, 322.5 > 122, 322.5 > 198; nonivamide 294.4 > 137, 294.4 > 122, 294.4 > 170). Product ion 137 was used for quantitation for capsaicin, and the other ions are used for qualification.
The trap column (Fig. 1(8)) was connected to the BPR to trap the extracts from the SFE vessel. And all the extracted compounds were trapped temporarily before the concentrator. After the BPR, SC-CO2 became CO2 gas because the pressure fell down and the supercritical conditions were not maintained. Then CO2 bubble was removed at the trap column. Three columns, GL Sciences ODS-3 (4.6 mm I.D. × 50 mm, particle size 10 mm), Shimadzu Shim-pack MAYI-ODS (4.6 mm I.D. × 50 mm, particle size 5 mm) and Shimpack VP-ODS (4.6 mm I.D. × 50 mm, particle size 5 mm), have been examined for the trap process. The three columns were compared and finally VP-ODS was selected as the trap column because of its long lifetime to the complex extracts.
Dilution line technology has been successfully applied to online enrichment for large volume injection [33-35]. In this system, the diluting line (Fig. 1(4, 9)) has been attached before the concentration column to reduce elution power from the modifier. Otherwise, the extracts would be directly flushed out of the concentration column by the modifier, which has a strong elution power.
Moreover, to reduce the elution power remarkably, higher polar solvent such as 100% water was used for the reverse phase liquid chromatography. In this case, SC-CO2 became CO2 gas at the trap column, and the modifier, methanol, remained to flush the extract into the concentration column. Addition of water through the dilution line increased the polarity of the mobile phase, and enhanced the retention of capsaicin on the concentration column.
Effect of methanol content in the mobile phase on the retention time of capsaicin on a C18 column was investigated. Retention time of capsaicin increased with decreasing the content of methanol in the mobile phase. When 25% methanol was used as the mobile phase, capsaicin was retained on the concentration column over 50 min. Furthermore, flow rate of the modifier, methanol, was fixed to be 0.5 mL/min, while flow rate of water in the dilution line was changed from 0.5 mL/min to 2.0 mL/min to obtain different polarity and elution ability. Fig. 2 shows the relationship between peak shape and the diluting line flow rate. Increasing flow rate of the dilution fluid (water) improved peak shape of the capsaicin effectively, indicating that capsaicin was strongly retained on the concentration column and concentrated to a narrow distribution in the column by using a higher flow rate of water. Since the peak shape obtained at 1.5 mL/min and 2.0 mL/min are both sharp and symmetric, a lower flow rate, 1.5 mL/min was selected for on-line concentration.

|
Download:
|
| Fig. 2. Relationship between diluting line flow rate and peak shape of capsaicinoids. (A) 2.0 mL/min; (B) 1.5 mL/min; (C) 1.2 mL/min; (D) 1.0 mL/min; (E) 0.5 mL/min. | |
{kind=link}
As discussed in the above section, after eluting from the trap column, the extract in methanol was mixed with water and then was carried into the concentration column for on-line enrichment (Fig. 1(4, 12)). High polar mobile phase was used to retain capsaicin strongly on the concentration column. A C18 guard column, 10 mm in length was selected as the concentration column to reduce the elution time after on-line enrichment had been accomplished. Band broadening, resulting from phase change of supercritical CO2 after BPR, was finally improved and capsaicin was effectively concentrated on the guard column.
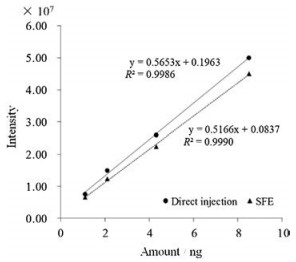
After optimization for the SFE-LCMS, a serial of standard solutions containing capsaicin (1.1 ng, 2.2 ng, 4.3 ng, 8.5 ng) were added into the SFE vessel. Then capsaicin in the standard solutions was on-line extracted and carried into the trap column by the supercritical CO2 fluid, modified by methanol. After on-line concentration, capsaicin standard was finally detected by mass spectrometry. Four points were employed to construct calibration curves (Fig. 3), which were of good linearity with a minimum coefficient of determination (R2) >0.998 in the range of 1.1–8.5 ng. Limit of detection (signal/noise = 3) for the capsaicin standard solution was estimated to be 0.55 pg.

|
Download:
|
| Fig. 3. Calibration curves of capsaicin: SFE and direct injection. | |
{kind=link}
Fig. 3 shows the calibration curves based on the SFE method and autosampler direct injection method. The peak areas detected for the SFE method were a little smaller than those measured based on the autosampler direct injection method. Recovery for the standard samples was calculated to be 90%.
Three types of bell peppers were on-line extracted and the extracts were separated and detected by MS/MS. Fig. 4 shows typical MS chromatogram of capsaicinoids in the green bell pepper. Capsaicin and dihydrocapsaicin, two major components, were detected at 10.10 min and 11.10 min, respectively. The two compounds were separated completely by using a reverse-phase C18 column. Synthetic capsaicin, nonivamide, was also detected at 10.10 min, was not separated from capsaicin. Since mass spectrometry was used for detection and only a small amount of nonivamide exists in natural foods, capsaicin can be determined by using this system. Moreover, although commercial standards of minor capsaicinoids were not available yet, the chromatogram of the extract shows a peak at 9.85 min (m/z 294.4 > 137.0), which could be putatively attributed to nordihydrocapsaicin, a peak at 11.35 min (m/z 320.4 > 137.0), which could be putatively attributed to homocapsaicin, and a peak at 12.45 min (m/z 322.5 > 137.0), that could be putatively attributed to homodihydrocapsaicin.

|
Download:
|
| Fig. 4. Typical MRM chromatograms of capsaicinoids in the green bell pepper. (A) All the capsaicinoids; (B) dihydrocapsaicin; (C) nonivamide; (D) homocapsaicin; (E) homodihydrocapsaicin; (F) nordihydrocapsaincin; (G) capsaicin (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article). | |
{kind=link}
Based on the calibration curve, capsaicin in green, yellow and red bell peppers was determined to be 60.33 ng/g, 31.79 ng/g and 35.38 ng/g, respectively, indicating the bell peppers measured in this study contain low content of capsaicin, which could not be detected in the previous studies [36, 37].
In summary, SFE was coupled with RPLC/MS for on-line extraction and reverse phase separation of fat-soluble compounds in complex matrices. This system was successfully used to direct determination of capsaicin in various capsicum fruits. It is expected to be applied to automated and rapid analysis of drug tablets and natural products without any tedious preparation.
AcknowledgmentThis work was supported by the National Natural Science Foundation of China (No. 21621003).
| [1] |
W. Frenzel, I. Markeviciute, J. Chromatogr. A 1479 (2017) 1-19. DOI:10.1016/j.chroma.2016.11.052 |
| [2] |
H.L. Qian, C.X. Yang, W.L. Wang, et al., J. Chromatogr. A 154 (2018) 1-18. |
| [3] |
Z. Wu, N. Xu, W. Li, et al., Chin. Chem. Lett. 30 (2019) 95-98. DOI:10.1016/j.cclet.2018.01.048 |
| [4] |
R.K. Saini, Y.S. Keum, Food Chem. 240 (2018) 90-103. DOI:10.1016/j.foodchem.2017.07.099 |
| [5] |
E. Reverchon, G.D. Porta, F. Senatore, J. Agric. Food Chem. 43 (1995) 1654-1658. DOI:10.1021/jf00054a045 |
| [6] |
E. Reverchon, G.D. Porta, J. Supercrit. Fluids 9 (1996) 199-204. DOI:10.1016/S0896-8446(96)90033-9 |
| [7] |
C. Turner, J.W. King, L. Mathiasson, J. Chromatogr. A 936 (2001) 215-237. DOI:10.1016/S0021-9673(01)01082-2 |
| [8] |
Z. Wang, Z. Cui, Chin. Chem. Lett. 27 (2016) 241-246. DOI:10.1016/j.cclet.2015.10.001 |
| [9] |
S.R. Rissato, M.S. Galhiane, F.R.N. Knoll, et al., J. Chromatogr. A 1048 (2004) 153-159. DOI:10.1016/S0021-9673(04)01213-0 |
| [10] |
M. Herrero, A. Cifuentes, E. Ibanez, Food Chem. 98 (2006) 136-148. DOI:10.1016/j.foodchem.2005.05.058 |
| [11] |
F. Sahena, I.S.M. Zaidul, S. Jinap, et al., J. Food Eng. 95 (2009) 240-253. DOI:10.1016/j.jfoodeng.2009.06.026 |
| [12] |
E. Reverchon, I. de Marco, J. Supercrit. Fluids 38 (2006) 146-166. DOI:10.1016/j.supflu.2006.03.020 |
| [13] |
S.M. Pourmortazavi, S.S. Hajimirsadeghi, J. Chromatogr. A 1163 (2007) 2-24. DOI:10.1016/j.chroma.2007.06.021 |
| [14] |
W.H. Chen, C.H. Chen, C.M.J. Chang, et al., J. Supercrit.Fluids 51 (2009) 174-180. DOI:10.1016/j.supflu.2009.08.010 |
| [15] |
J.A. Wood, M.A. Bernards, W.K. Wan, et al., J. Supercrit. Fluids 39 (2006) 40-47. DOI:10.1016/j.supflu.2006.01.016 |
| [16] |
I. Pasquali, R. Bettini, Int. J. Pharm. 364 (2008) 176-187. DOI:10.1016/j.ijpharm.2008.05.014 |
| [17] |
M. Herrero, J.A. Mendiola, A. Cifuentes, et al., J. Chromatogr. A 1217 (2010) 2495-2511. DOI:10.1016/j.chroma.2009.12.019 |
| [18] |
J. Sunarso, S. Ismadji, J. Hazard. Mater. 161 (2009) 1-20. DOI:10.1016/j.jhazmat.2008.03.069 |
| [19] |
M. Zoccali, D. Giuffrida, P. Dugo, et al., J. Sep. Sci. 40 (2017) 3905-3913. DOI:10.1002/jssc.v40.19 |
| [20] |
D. Giuffrida, M. Zoccali, A. Arigò, et al., Arch. Biochem.Biophys. 646 (2018) 161-167. DOI:10.1016/j.abb.2018.03.011 |
| [21] |
Y. Liang, J. Liu, Q. Zhong, et al., J. Sep. Sci. 41 (2018) 1600-1609. DOI:10.1002/jssc.v41.7 |
| [22] |
K. Suto, Y. Ito, K. Sagara, et al., J. Chromatogr. A 786 (1997) 366-370. DOI:10.1016/S0021-9673(97)00592-X |
| [23] |
X. Zhang, F. Ji, Y. Li, et al., Anal. Sci. 34 (2018) 407-413. DOI:10.2116/analsci.17P434 |
| [24] |
M. Suzuki, S. Nishiumi, T. Kobayashi, et al., Rapid Commun. Mass Spectrom. 31 (2017) 886-894. DOI:10.1002/rcm.7857 |
| [25] |
D. Okamoto, Y. Hirata, Anal. Sci. 22 (2006) 1437-1440. DOI:10.2116/analsci.22.1437 |
| [26] |
A.P. Wicker, D.D. Carlton, K. Tanaka, et al., J. Chromatog. B 1086 (2018) 82-88. DOI:10.1016/j.jchromb.2018.04.014 |
| [27] |
K.K. Unger, P. Roumeliotis, J. Chromatogr. A 282 (1983) 519-526. DOI:10.1016/S0021-9673(00)91629-7 |
| [28] |
J. Zhang, L. Zhang, J. Duan, et al., J. Sep. Sci. 29 (2006) 2514-2522. DOI:10.1002/(ISSN)1615-9314 |
| [29] |
E.D. Ramsey, A.T. Rees, G. Wei, et al., J. Chromatogr. A 1217 (2010) 3348-3356. DOI:10.1016/j.chroma.2010.03.025 |
| [30] |
M. Ashraf-Khorassani, N. Nazem, L.T. Taylor, J. Chromatogr. A 995 (2003) 227-232. DOI:10.1016/S0021-9673(03)00520-X |
| [31] |
Z. Wang, M. Ashraf-Khorassani, L.T. Taylor, J.ChromatogrJ. Chromatogr. A 1033 (2004) 221-227. DOI:10.1016/j.chroma.2004.01.041 |
| [32] |
N. Hamada, Y. Guo, F. Ji, et al., Talanta 190 (2018) 9-14. DOI:10.1016/j.talanta.2018.07.063 |
| [33] |
S.I. Kawano, H. Murakita, E. Yamamoto, et al., J. Chromatogr. B 792 (2003) 49-54. DOI:10.1016/S1570-0232(03)00309-X |
| [34] |
M. Liu, Y. Hashi, F. Pan, et al., J. Chromatogr. A 1133 (2006) 142-148. DOI:10.1016/j.chroma.2006.08.009 |
| [35] |
Y. Hashi, T.R. Wang, W. Du, et al., Talanta 74 (2008) 986-991. DOI:10.1016/j.talanta.2007.08.011 |
| [36] |
Z.A. Alothman, S.M. Wabaidur, M.R. Khan, et al., J. Sep. Sci. 35 (2012) 2892-2896. DOI:10.1002/jssc.v35.21 |
| [37] |
A. Garcés-Claver, M.S. Arnedo-Andrés, J. Abadía, et al., J. Agric. Food Chem. 54 (2006) 9303-9311. DOI:10.1021/jf0620261 |