 2019, Vol. 30
2019, Vol. 30 

 ,
Qing Zhua,*
,
Qing Zhua,*
b Department of Chemistry, National University of Singapore, Singapore 117543, Singapore;
c Key Laboratory of Flexible Electronics(KLOFE) & Institute of Advanced Materials(IAM), Jiangsu National Synergetic Innovation Center for Advanced Materials(SICAM), Nanjing Tech University(Nanjing Tech), Nanjing 211816, China
Phosphoinositide 3-kinases (PI3Ks), as a family of lipid kinases, are crucial regulators in a wide range of cellular processes [1-3]. The class Ⅰ PI3Ks has been most widely characterized for oncogenic properties for decades. 30% of human cancers including lung and malignant melanoma contain somatic mutations in PIK3CA gene, which encodes p100α catalytic subunit of PI3Ks [4]. Hence, they have consequently been recognized as potential drug targets [5-7]. Wortmannin (WM), as a known potent inhibitor of PI3K is a furanosteroid metabolite isolated from the fungi Penicillium funiculosum and Talaromyces (Penicillium) wortmannii [8]. It is extensively used in studies on endosomal organization and trafficking. It targets PI3Ks with an IC50 of approximately 5 nmol/L, and other proteins, such as DNA-dependent protein kinase (DNA-PK) with a dose-dependent manner [9-11]. Its inhibition mechanism against PI3Ks is shown in Fig. 1. The furan ring of WM is opened by the covalent reaction between C20 atom and the amine of a lysine in the ATP binding site of the catalytic subunit of PI3Ks to form an additional six-member ring with an intramolecular hydrogen bond, causing irreversible inhibition of PI3Ks activity [12-14]. However, although C20 atom provides the desired property, its inherent electrophilic character renders it susceptible to be attacked by other nucleophiles nearby, including thiol, primary and secondary amine in side chains of amino acids, leading to the high instability and toxicity of WM [15].

|
Download:
|
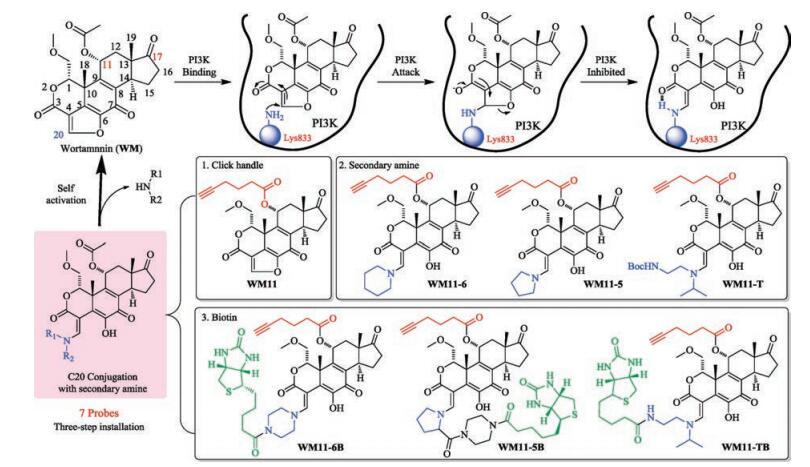
| Fig. 1. General mechanism of inhibition of PI3K by wortmannin (WM) and tertiary amine-C20 conjugated prodrugs. The structures of 7 probes were shown in the figure with three-step modifications. | |
{kind=link}
Many efforts were placed to reconcile the paradoxical fact that WM, although unstable, can yield high inhibition to PI3Ks [16-19]. Studies have revealed that the ring-opened virdin modifications at C20 position could bring significant pharmacological improvements [17, 18], such as PX-866, a potent and irreversible inhibitor against PI3Ks (IC50 = 0.1~88 nmol/L) [19]. As illustrated in Fig. 1, WM-conjugates involving tertiary enamine attached to C20 can slowly self-activate through intramolecular attack by C6 hydroxyl group to generate active WM in situ which can subsequently inhibit PI3Ks. Such prodrug modifications can produce locally high concentrations of WM in vivo to allow superior inhibition [18]. But still there is short of information about their cellular effects.
Activity-based probe profiling (ABPP), by making use of the socalled activity-based probes (ABPs)that were derived from bioactive compounds, is a powerful tool capable of identification of potential protein targets in a native biological setting [20-23]. Introduction of a simple modification onto the lead molecule, such as an alkyne tag, is a way to not only minimize the effect to its pharmacodynamic properties but also use a click-based in situ profiling approach to unveil targets [24, 25]. Previously, we had applied this strategy to successfully profile new targets of a vinyl-sulfone based compounds [26] and label protein tyrosine phosphatases using multicomponent Ugi reaction [27]. Here we extend this strategy to build WM based activity-based probes.
Considering improvement with the tertiary enamine selfactivating process, we were motivated to explore their effects in cellular systems, including cell proliferation and PI3K/Akt pathway. Herein, to accomplish this goal, we fused tertiary enaminebased WM derivatives together with ABPP approach to demonstrate their cellular effects. Furthermore, we also equipped a cancer-targeting [28] vitamin-biotin on C20 site of WM. Biotin is specific to the vitamin receptors overexpressed on tumor cell surface and has been used as a drug delivery tool allowing drug conjugates to be internalized efficiently through receptor-mediated endocytosis [28-30]. Hence, seven compounds shown in Fig. 1 were synthesized and further studied using different assays with comparison to their parent WM. We found IC50 of self-activating probes are better than WM. Biotin attachment could improve the cancer cell uptake. Furthermore, as the activation time is dependent on the amine attached, WM and its derivatives could decrease and block Akt phosphorylation at a short time but with different prolonged turn-over time. Finally, protein profiling and pull-down experiments showed the probes have numerous targets including labeling different PI3K catalytic subunits with selectivity.
Structural modifications at either C11 or C17 of WM have been reported. Changes at C11 site did not significantly alter its native biological activities [31-34]. However, bulky fluorophore or biotin modification at C11 could affect its binding affinity [32]. We took advantage of this key feature in the design of our activity-based probes. 1) The first step is to introduce a clickable alkyne handle to the alcohol group of C11 site through an ester linkage, giving a probe WM11. 2) Then, three common secondary amines, piperidine, proline and asymmetric bulky amine were installed at C20 to form three self-activating probes, providing the corresponding WM11-6/WM11-5/WM11-T, respectively. 3) To achieve better cellpermeability and cancer selectivity, a cancer-targeting ligand, biotin, was installed into the amine portion to enhance cancer cell uptake, obtaining WM11-6B/WM11-5B/WM11-TB, respectively. The synthesis of all the probes was commenced from the original WM and the yield of each step was quantitative. The detailed synthetic schemes and procedures were shown in Supporting information. All compounds were characterized by NMR and HRMS.
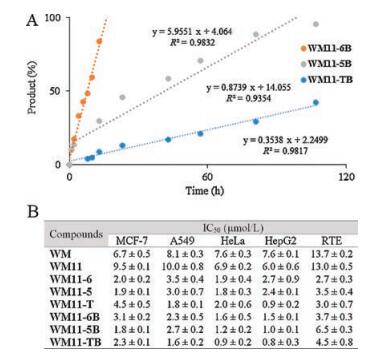
The secondary amine-modified WM would undergo exchange reaction with primary amine to form more stable products as shown Fig. 1. Therefore, with the compounds in hand, we first examined the self-activation of three amine modifications using HPLC. 500 μmol/L WM11-6B, WM11-5B and WM11-TB were incubated with benzyl amine (2.5 μmol/L, 50 equiv.) in water/ ACN (1:1, v/v). The reactions were monitored by HPLC at different time points. As shown in Fig. 2 and Fig. S1 (Supporting information), all of them showed a progressive increase in the formation of benzyl amine-added products and decrease in the starting probes with time. The reaction with WM11-6B was finished in 16 h, while, the rate of WM11-5B and WM11-TB were slow, around 100 h and 260 h, respectively. Hence, the tertiary amine in WM derivatives, temporally deactivating WM reactivity, served as a leaving group, which is critical to WM activation.

|
Download:
|
| Fig. 2. (A) Linearized curve of the reactions between WM derivatives and benzyl amine using HPLC analysis. Each time point was calculated and analyzed using the peak area of starting materials at 405 nm. (B) IC50 value of WM and 7 probes against 5 cell lines. | |
{kind=link}
Next, we evaluated their cellular activities by measuring the IC50 (inhibition of 50% cell growth) values of probes against several different normal and cancer cell lines. The antiproliferation assay was performed after 72 h incubation of probes using the tetrazolium dye MTT. Due to the close absorption wavelength of furan ring-opened structures (λabs = 420 nm, yellow, Fig. S2 in Supporting information) of WM and the formazan (λabs = 450 nm, yellow) reduced from XTT reagent, we chose MTT, which is reduced to purple formazan (λabs = 570 nm) in living cells, to screen for cell viability to provide more accurate results. Fig. 2 and Fig. S4 in Supporting information are the summary of IC50 of 8 compounds against MCF-7, A549, HeLa, HepG2 and RTE cell lines. 7 probes and WM showed a dose-dependent inhibition of all kinds of cell lines over a 72 h-time period. According to the results, the installation of alkyne handle on the C11 does not alter the inhibitory effect significantly. All probes with self-activating property demonstrated several-fold higher potency, especially WM11-T showing IC50 value in the nanomolar range (around 900 nmol/L against HepG2), whereas that of WM was found to be around 10 μmol/L [18]. This prominent improvement could be attributed to their improved chemical stability through a transient ring-opened form. Furthermore, with cancer-targeting reagent-biotin, all three biotinylated probes, including WM11-6B, WM11-5B and WM11-TB, 1.6 μmol/L, 1.2 μmol/L and 0.9 μmol/L against HeLa, respectively, showed even higher potency than those without biotin. However, this increment was not obviously observed in RTE cell line, a normal cell line. It is apparent that the internalization of prodrugs was enhanced by biotin in cancer cells. Overall, it is reasonable to conclude that both self-activation and biotin conjugation generally enhanced the inhibitory property of the parent WM.
According to cell proliferation assay, the prodrugs showed better inhibitory properties than WM. WM is a known PI3K pathway inhibitor as the oncogenic PI3Ks regulate Akt activation phosphorylation at Thr308. The degree of phosphorylation of Akt at Thr308 in response to the probes was then determined by western blotting with phospho-Akt (Thr308) under different treatments [1, 5]. We selected 250 nmol/L to screen all compounds against pAkt after analysis of WM11 and WM11-5 in different conditions (Fig. S5 in Supporting information). As shown in Fig. 3A, WM11-T and WM11-TB, with better cellular toxicity and slow selfactivation showed less influence on Akt phosphorylation. Other probes showed significant inhibition upon treatment to nearly complete suppression of the phosphorylation of both sites under 4 h incubation, indicating there was a direct and fast interaction between probes and PI3Ks. WM11-5 showed slower inhibition than those with piperazine, suggesting slow rate of self-activation of WM11-5 may affect the binding with PI3Ks. Through comparison between WM11-5 and WM11-5B, it seems that uptake of WM11-5B, with the assistance of biotin, could accelerate the inhibition process, but the recovery of phosphorylation still occurred at 20 h incubation. Overall, after 8 h incubation, most of probes ceased to inhibit the phosphorylation and the Akt phosphorylation was fully turned over in 24 h, assuming activation of Akt-independent PI3K pathways or negative feedback loops may occur [35-37]. Taken together, these data suggested that most of inhibitors could decrease the phosphorylation of Akt under lower concentrations, especially self-activating prodrug approach, through slow release to enhance the effectiveness.

|
Download:
|
| Fig. 3. (A) Cells were treated with 250 nmol/L WM or its analog at indicated time. Cells were directly subject to loading dye and immunoblot analysis for pAkt (Thr308, respectively). Anti-Akt as loading controls (partially were shown in the figure). (B) The pull-down (PD) gels of A549 cells labelled with the corresponding probes (10 μmol/L, 4 h). (C) Four PI3K catalytic subunit-target validation of pulldown samples from B). | |
{kind=link}
Since the inhibition to Akt phosphorylation is not persistent, we further explored their capability to label PI3Ks in cellular systems. To demonstrate this, we took advantage of the clickable alkyne handle in the design of our probes to investigate their PI3Ks binding properties through the biorthogonal click chemistry. Probes (10 μmol/L) were directly added into A549 cell culture medium. After indicated different time (4 h, 8 h and 20 h), cells were washed to remove excessive probes, homogenized, clicked with TER-N3 under Cu-catalyzed click-chemistry (CuAAC) conditions, separated by SDS-PAGE gel, and analysed by in-gel fluorescence scanning. According to the working mechanism of WM, WM could reversibly react with thiol in cysteine and secondary amine in proline, but irreversibly bind with the side chain of lysine at the C20 position on furan ring [15]. So, it is interesting to note that the reversible binding could be to some degree abolished by Tris in SDS loading dye (Fig. S3), suggesting only irreversible labelled proteins by our probes could be visualized. As shown in Fig. S6 (Supporting information), the labelling patterns are similar among all probes with many protein targets [33, 34], whereas WM11-T and WM11-TB consistently produced weaker labelled bands due to their slow activation process. WM11, with no requirement of the activation step, showed stronger labelling efficiency than other probes. The labelling intensity decreased after longer incubation mainly due to the hydrolysis of ester bond between the alkyne tag and WM moiety. Nevertheless, we observed several highly distinctive bands at around 35-55 kD, but only faint bands around 110-125 kD as we expected for the catalytic subunits of PI3Ks.
Hence, to more directly probe their binding affinity toward PI3Ks in live cells, we continued to enrich the labelled protein extract by large scale pulldown using avidin-agarose beads, separated by SDS-PAGE gel, detected by western blotting using four different PI3K antibodies (p110α, p110β, p110γ, Vps34). As bulky WM11-T and WM11-TB could not show stronger labelling, we analyzed the rest five probes. In situ proteome pull-down results and western blot were shown in Figs. 3B and C, respectively. WM11 showed better in situ subunits labeling, presumably because of its faster reactivity. Similar targeting patterns from other four probe-treated cells were obtained. From Fig. 3C, three in four subunits, including p110α, p110β and Vps34, were indeed successfully detected in probe-labeled cells but not in cells treated with DMSO. Interestingly, PI3K class IB, p110γ, which bind Lys 833 site of p110γ in vitro [38], was minimally labelled by probes. And class Ⅲ catalytic subunit Vps34, on the other hand, was positively labelled by all the probes. Therefore, these results indicated that WM and derivatives indeed have labelling affinity toward PI3Ks, but under cellular condition, not like in vitro assays, the efficiency is weak and with selectivity in some degrees.
In conclusion, we have synthesized different self-activating prodrug WM probes. These derivatives demonstrated promising improvement of cytotoxic activity against several cancer cell lines compared with the original. Furthermore, this series of derivatives displayed high inhibition toward Akt phosphorylation with prolonged turn-over time. These WM-based ABPP probes were used for profiling their targets in living cells. Our finding indicates WM and derivatives are highly reactive compounds and engaged in covalent interactions with numerous targets in cellular system, possible the major reason causing high toxicity. They indeed bind to PI3K catalytic subunits with selectivity and low degree which could be one of reasons causing the unsustainable inhibition to Akt phosphorylation. Notwithstanding, we still caution this bioactive compound should be further investigated their functions in PI3K and their mechanism in cellular systems as a potential drug candidate. Furthermore, we believe the use of activity-based protein profiling (in situ drug profiling) is becoming an efficient tool in understanding its mode of action and origin of side effects due to off targets binding [39, 40].
AcknowledgmentsThis work is financially supported by the Natural Science Foundation of Zhejiang Province (Nos. LQ16B020003, LY17B060009), the National Natural Science Foundation of China (Nos. 21708034, 21472172, 81672508, 61505076) and Jiangsu Provincial Foundation for Distinguished Young Scholars (No. BK20170041). We thank Prof. Shao Q. Yao (National University of Singapore) for his invaluable support for this work.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2018.05.030.
| [1] |
B. Vanhaesebroeck, L. Stephens, P. Hawkins, Nat. Rev. Mole. Cell. Biol. 13 (2012) 195-203. DOI:10.1038/nrm3290 |
| [2] |
M.P. Wymann, C. Schultz, ChemBioChem 13 (2012) 2022-2035. DOI:10.1002/cbic.v13.14 |
| [3] |
D.A. Fruman, H. Chiu, B.D. Hopkins, et al., Cell 170 (2017) 605-635. DOI:10.1016/j.cell.2017.07.029 |
| [4] |
S. Jean, A.A. Kiger, J. Cell. Sci. 127 (2014) 923-928. |
| [5] |
B.T. Hennessy, D.L. Smith, P.T. Ram, et al., Nat. Rev. Drug. Discov. 4 (2005) 988-1004. DOI:10.1038/nrd1902 |
| [6] |
C. Massacesi, E. Di, P. Tomaso, Urban, et al., Onco. Targets. Ther. 9 (2016) 203-210. |
| [7] |
G.R. Gao, J.L. Liu, D.S. Mei, et al., Chin. Chem. Lett. 26 (2015) 118-120. DOI:10.1016/j.cclet.2014.10.016 |
| [8] |
P.W. Brian, P.J. Curtis, H.G. Hemming, et al., Trans. Brit. Mycol. Soc. 40 (1957) 365-368. DOI:10.1016/S0007-1536(57)80033-3 |
| [9] |
A. Arcaro, M.P. Wymann, Biochem. J. 296 (1993) 297-301. DOI:10.1042/bj2960297 |
| [10] |
J.N. Sarkaria, R.S. Tibbetts, E.C. Busby, et al., Cancer. Res. 58 (1998) 4275-4382. |
| [11] |
H. Yano, S. Nakanishi, K. Kimura, et al., J. Biol. Chem. 268 (1993) 25846-25856. |
| [12] |
P. Wipf, R.J. Halter, Org. Biomol. Chem. 3 (2005) 2053-2061. DOI:10.1039/b504418a |
| [13] |
E.H. Walker, M.E. Pacold, O. Perisic, et al., Mol. Cell. 6 (2000) 909-919. DOI:10.1016/S1097-2765(05)00089-4 |
| [14] |
M.P. Wymann, G. Bulgarelli-Leva, M.J. Zvelebil, et al., Mol. Cell. Biol. 16 (1996) 1722-1733. DOI:10.1128/MCB.16.4.1722 |
| [15] |
H. Yuan, K.R. Barnes, R. Weissleder, L. Cantley, L. Josephson, Chem. Biol. 14 (2007) 321-328. DOI:10.1016/j.chembiol.2007.02.007 |
| [16] |
S. Karve, M.E. Werner, R. Sukumar, et al., Proc. Natl. Acad. Sci. U. S. A. 109 (2012) 8230-8235. DOI:10.1073/pnas.1120508109 |
| [17] |
A. Smith, J. Blois, H. Yuan, et al., Mol. Cancer. Ther. 8 (2009) 1666-1675. DOI:10.1158/1535-7163.MCT-08-1012 |
| [18] |
P. Wipf, D.J. Minion, R.J. Halter, et al., Org. Biomol. Chem. 2 (2004) 1911-1920. DOI:10.1039/b405431h |
| [19] |
N.T. Ihle, R. Williams, S. Chow, et al., Mol. Cancer. Ther. 3 (2004) 763-772. |
| [20] |
W.P. Heal, T.H.T. Dang, E.W. Tate, Chem. Soc. Rev. 40 (2011) 246-257. DOI:10.1039/C0CS00004C |
| [21] |
Y. Su, J. Ge, B. Zhu, et al., Curr. Opin. Chem. Biol. 17 (2013) 768-775. DOI:10.1016/j.cbpa.2013.06.005 |
| [22] |
P. Yang, K. Liu, ChemBioChem 6 (2015) 712-724. |
| [23] |
W. Wang, D.S. Tekcham, M. Yan, et al., Chin. Chem. Lett. 29 (2018) 645-647. DOI:10.1016/j.cclet.2017.10.002 |
| [24] |
P. Yang, K. Liu, M. Ngai, et al., J. Am. Chem. Soc. 132 (2010) 656-666. DOI:10.1021/ja907716f |
| [25] |
S. Pan, H. Zhang, C. Wang, S.C.L. Yao, S.Q. Yao, Nat. Prod. Rep. 33 (2016) 612-620. DOI:10.1039/C5NP00101C |
| [26] |
J. Ge, C.J. Zhang, L. Li, et al., ACS. Chem. Biol. 8 (2013) 2577-2585. DOI:10.1021/cb4002602 |
| [27] |
J. Ge, X. Cheng, L.P. Tan, S.Q. Yao, Chem. Commun. 48 (2012) 4453-4455. DOI:10.1039/c2cc31294h |
| [28] |
S. Jaracz, J. Chen, L.V. Kuznetsova, I. Ojima, Bioorg. Med. Chem. 13 (2005) 5043-5054. |
| [29] |
S. Maiti, N. Park, J.H. Han, et al., J. Am. Chem. Soc. 135 (2013) 4567-4572. DOI:10.1021/ja401350x |
| [30] |
S. Chen, X. Zhao, J. Chen, et al., Bioconjugate. Chem. 21 (2010) 979-987. DOI:10.1021/bc9005656 |
| [31] |
A. Zask, J. Kaplan, L. Toral-Barza, et al., J. Med. Chem. 51 (2008) 1319-1323. DOI:10.1021/jm7012858 |
| [32] |
M.C. Yee, S.C. Fas, M.M. Stohlmeyer, et al., J. Biol. Chem. 280 (2005) 29053-29059. DOI:10.1074/jbc.M504730200 |
| [33] |
Y. Liu, K.R. Shreder, W. Gai, et al., Chem. Biol. 12 (2005) 99-107. DOI:10.1016/j.chembiol.2004.11.009 |
| [34] |
G.F. Desrochers, A.R. Sherratt, D.R. Blais, et al., ACS. Infect. Dis. 1 (2015) 443-452. DOI:10.1021/acsinfecdis.5b00083 |
| [35] |
M. Dufour, A. Dormond-Meuwly, C. Pythoud, et al., Biochem. Biophys. Res. Commun. 438 (2013) 32-37. |
| [36] |
K. Mahajan, N.P. Mahajan, J. Cell. Physiol. 227 (2012) 3178-3184. DOI:10.1002/jcp.24065 |
| [37] |
B.D. Manning, A. Toker, Cell. 169 (2017) 381-405. DOI:10.1016/j.cell.2017.04.001 |
| [38] |
S. Stoyanova, G. Bulgarelli-Leva, C. Kirsch, et al., Biochem. J. 324 (1997) 489-495. DOI:10.1042/bj3240489 |
| [39] |
A.R. Sherratt, N. Nasheri, C.S. Mckay, et al., ChemBioChem 15 (2014) 1253-1256. DOI:10.1002/cbic.v15.9 |
| [40] |
L. Li, H. Wijaya, S. Samanta, Y. Lam, S.Q. Yao, Sci. Rep. 5 (2015) 11522-11530. DOI:10.1038/srep11522 |