 2019, Vol. 30
2019, Vol. 30 


Over the past few decades, donor-acceptor (D-A) conjugated polymers [1, 2] have been promising candidates for low-cost, flexible and large-scale engineering materials [3, 4] of the next-generation electronics [5], due to their solution-processability and good mechanic properties. D-A conjugated polymers usually consist of two parts: the donor units and the acceptor units. The acceptor units have great impact on the lowest unoccupied molecular orbital (LUMO) energy levels, as well as the molecular conformation. To the best of our knowledge, there have been many electron-deficient aromatic units such as isoindigo (IID) [6-8], diketopyrrolopyrrole (DPP) [9, 10], benzothiadiazole (BT) [11-13], and naphthalene diimide (NDI) [14, 15], which are proved to have the potential to enhance the optoelectronic properties of conjugated polymers. Although a number of acceptor units have been rapidly developed, strategies to construct high-performance acceptor units are still limited.
A common strategy to achieve electron-deficient acceptors is introducing electron-withdrawing groups (such as F, Cl, CN) onto the conjugated backbones to lower their LUMO levels [16-18]. Besides this, embedding sp2-nitrogen in benzene ring to form pyridine ring in acceptors [19-21] can also further lower the energy levels of the acceptor moiety of D-A conjugated polymers. Swager et al first developed a promising class of water and/or methanol soluble n-type poly(pyridinium phenylene)s [1, 2], which may find utility in photovoltaic devices when blended with donor conjugated polymers. Introduction of electron-withdrawing sp2-nitrogen groups onto molecular backbones [22-24] may not only tune the frontier molecular orbital energy levels of the polymers, but also lock the conformation of conjugated polymer backbones through intramolecular noncovalent interactions [25], which is beneficial to charge transport [26]. In this review, we summarize the recent development of D-A conjugated polymers with acceptors containing pyridine units, which mainly relates to three types of acceptors: azaisoindigos (7DNIID, 5DNIID), azaBDOPV (AzaBDOPV), and pyridal[2, 1, 3]thiadiazole (PT), as shown in Fig. 1.

|
Download:
|
| Fig. 1. Molecular structures of electron-deficient acceptors containing pyridine units. | |
{kind=link}
2. D-A conjugated polymers based on azaisoindigo 2.1. Design of azaisoindigo-based conjugated polymers
Recently, isoindigo has been extensively explored as electron-accepting moiety in organic field-effect transistors (OFETs) and organic light-emitting diodes (OLEDs) materials [6, 27, 28]. According to the calculation results, isoindigo has a torsion angle of ca. 20°-40° due to C-H⋯O=C steric interactions between the phenyl ring and the neighboring moiety [29] (Fig. 2B). In order to improve the conjugation of isoindigo, several strategies have been designed to enhance the coplanarity [30]. Ashraf and coworkers replaced the benzene units in isoindigo with thiopheneunits to decrease the steric repulsion [31]. Introducing thiophene units into isoindigo is favorable for efficient charge transport due to the increased planarity. The resulted new isoindigo derivative was applied to copolymers, which showed both good hole and electron mobilities up to 0.1 cm2 V-1 s-1. Afterwards, this strategy is extensively explored by Wan and Jassen to construct conjugated polymers for electronics [32, 33]. Moreover, fluorinated isoindigos having more planar conformation were proved by Pei and others [34-36] using the "conformational lock" strategy.

|
Download:
|
| Fig. 2. (A) Molecular structures and (B) the optimized structures and energy levels of isoindigo derivatives according to density functional theory at the B3LYP/6-31G* level. | |
{kind=link}
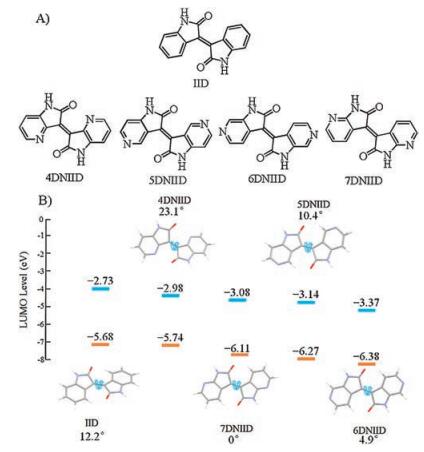
To achieve high-performance electronic materials, there is a growing demand to develop new strategies to achieve planar isoindigo derivatives. Inspired by the application of azaisoindigo in medicinal chemistry [37], Pei and Yu developed several azaisoindigo-based D-A conjugated polymers, respectively. The conformation and energy levels of these azaisoindigo units were calculated to elucidate the influence of the different nitrogen positions on the properties of azaisoindigos. According to the computational results (Fig. 2B), the different positions of nitrogen atoms in azaisoindigos obviously influence their planarity and LUMO levels. Compared to isoindigo, azaisoindigos with nitrogen at 5, 6, 7 positions show more planar conformation with torsion angles of 10.4°, 4.9° and 0° in 5, 5′-diazaisoindigo (5DNIID), 6, 6′-diazaisoindigo (6DNIID) and 7, 7′-diazaisoindigo (7DNIID), respectively. Notably, all the azaisoindigos show lower LUMO levels as compared to isoindigo, indicating that embedding sp2-nitrogen in benzene ring to form pyridine ring is indeed an effective strategy to develop new electron-deficient acceptors. Several synthetic routes have been designed to synthesize azaisoindigo and their derivatives, which usually involve brominaton and oxidation reactions [38-40]. However, because of the high reactivity of 4DNIID and 6DNIID, only 5- and 7-azaisoindigos were explored in D-A conjugated polymers [29, 40-42].
In 2016, Yu et al. synthesized two D-A conjugated polymers, PAIID-BT-C1andPAIID-BT-C3, with 7DNIID as acceptor and bithiophene as donor [29], and fabricated OFETs based on these polymers with top-gate/bottom-contact (TGBC) and bottom-gate/bottome-contact (BGBC) configurations through spin-coating. They presented that PAIID-BT-C3 (Fig. 3A) achieved hole mobilities as high as 7.28 cm2 V-1 s-1 in BGBC configuration and ambipolar mobilities of 2.33/0.78 cm2 V-1 s-1 in TGBC configuration, indicating that PAIID-BT-C3 is an excellent organic bipolar semiconducting material.

|
Download:
|
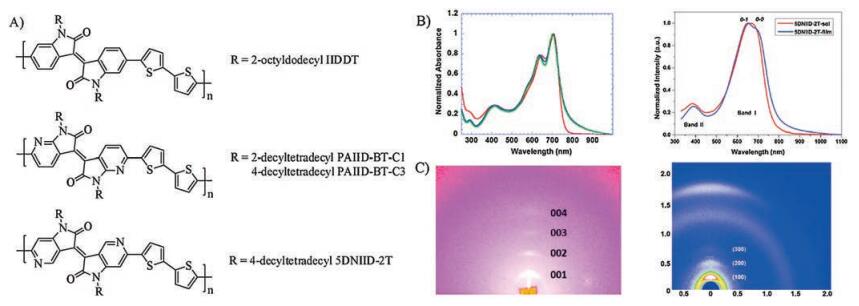
| Fig. 3. A) Molecular structures of D-A conjugated polymers based on azaisoindigos; B) UV-vis-NIR absorption spectra of IIDDT (left) and 5DNIID-2T (right); Copied with permission for IIDDT from Ref [6]. Copyright 2011, American Chemical Society. C) 2D-GIXD patterns of IIDDT (left) and 5DNIID-2T (right). Copied with permission for 5DNIID-2T from Ref [40]. Copyright 2017, Wiley-VCH. | |
{kind=link}
In 2017, Pei et al. reported the first example of D-A conjugated polymer based on 5DNIID. They successfully copolymerized 5DNIID with bithiophene unit to construct a D-A polymer 5DNIID-2T (Fig. 3A) and demonstrated its application in OFETs [40]. Compared with the isoindigo-based conjugated polymer IIDDT, 5DNIID-2T exhibited a lower LUMO level as expected.
The molecular weights (Mn) of IIDDT [6] and 5DNIID-2T were measured to be 33.7 kDa and 33.46 kDa, respectively, by high-temperature gel permeation chromatography (GPC) using 1, 2, 4-trichlorobenzene as the eluent at 150 ℃. The UV-vis spectra of both polymers (IIDDT, 5DNIID-2T) showed two characteristic bands (Fig. 3B), where the high-energy band from 300 nm to 450 nm was attributed to the π-π* transition of the IID and 5DNIID units, and the low-energy band from 450 nm to 900 nm originated from the intramolecular charge transfer (ICT) absorption. In addition, grazing incidence X-ray diffraction (GIXD) was used to investigate polymer packing in film (Fig. 3C). IIDDT showed four diffraction peaks in out-of-plane direction with a strong diffraction peak at 2θ = 3.58°, corresponding to a d-spacing of 19.88 Å (λ = 1.240 Å). However, 5DNIID-2T showed a weaker diffraction peak at 2θ = 3.00°, which corresponds to a d-spacing of 23.68 Å (λ = 1.2398 Å). Two other weak diffraction peaks were also observed in 5DNIID-2T film which were ascribed to (200) and (300) diffractions. Different from the crystalline fibrillar intercalating network of IIDDT film [6], the film of 5DNIIT-2T was more amorphous as shown in the tapping-mode atomic force microscopy(AFM) images. As a result, 5DNIID-2T showed lower carrier mobility than IIDDT. Further chemical modification of 5DNIID-2T might provide the opportunity to achieve better charge transport properties. In addition, 5DNIID-2T showed obvious different charge transport behaviors in OFETs with PAIID-BT-C3, which may be related to the different device configurations.
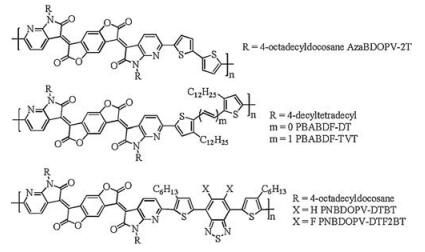
2.2. Structure-property relationships of AzaBDOPV-based conjugated polymersIn 2016, Pei et al. first embedded sp2-nitrogen atoms into benzodifurandione-based oligo(p-phenylene vinylene) (BDOPV) backbone [41] to construct a new acceptor AzaBDOPV, and developed a D-A conjugated polymer AzaBDOPV-2T (Fig. 4), which showed high electron mobility of 3.22 cm2 V-1 s-1 under ambient conditions.

|
Download:
|
| Fig. 4. Molecular structures of D-A conjugated polymers based on AzaBDOPV. | |
{kind=link}
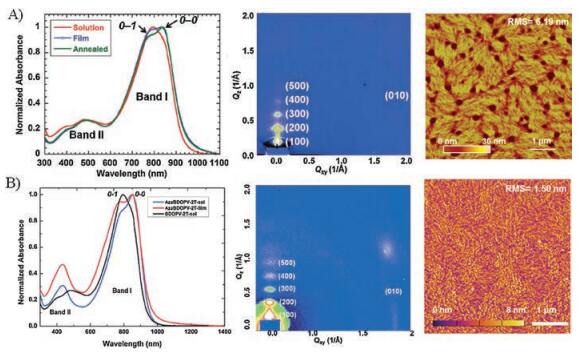
Compared with BDOPV-2T, AzaBDOPV-2T exhibited a more planar backbone and lower LUMO level of -4.37 eV (Fig. 5), which could be inferred from the absorption spectra. The absorption spectra of both BDOPV-2T and AzaBDOPV-2T displayed dual-band absorption and obvious 0-0 and 0-1 vibrational peaks both in solution and in films. However, the 0-0 vibrational peak of AzaBDOPV-2T was more obvious in solution, suggesting a more planar conformation. GIXD and tapping-mode AFM experiments indicated a distinct edge-on lamellar packing and stronger crystallinity in AzaBDOPV-2T films, which could be attributed to stronger interchain interactions and more ordered microstructures. These results demonstrated that AzaBDOPV was an excellent acceptor for constructing high-performance D-A conjugated polymers.

|
Download:
|
| Fig. 5. UV-vis-NIR spectrum, 2D-GIXD patterns and AFM images of BDOPV-2T (A) and AzaBDOPV-2T (B). Reproduced with permission [43]. Copyright 2013, Wiley-VCH. | |
{kind=link}
In 2017, Cho and his coworkers applied alkylated bithiophene and (E)-2-(2-(thiophen-2-yl)vinyl)thiophene as donor unit [42] to synthesize two AzaBDOPV-based conjugated polymers PBABDF-DT and PBABDF-TVT (Fig. 4). The LUMO levels of PBABDF-DT and PBABDF-TVT were -4.04 eV and -3.99 eV according to the cyclic voltammetry experiments, which were 0.34 eV and 0.29 eV lower than that of BDOPV-2T, but higher than that of AzaBDOPV-2T. OFETs were fabricated with bottom-gate/top-contact (BGTC) configuration through spin-coating. These two polymers displayed electron mobilities up to 1.86 cm2 V-1 s-1 and 1.56 cm2 V-1 s-1 after annealing process. Grazing incident X-ray diffraction showed the π-π stacking distance of PBABDF-DT and PBABDF-TVT were 3.52 Å and 3.50 Å, respectively. The relatively higher LUMO levels and larger π-π distance might result in the poorer performance of these two polymers than that of AzaBDOPV-2T. Recently, Yu et al. reported two ambipolar conjugated polymers, PNBDOPV-DTBT and PNBDOPV-DTF2BT, based on AzaBDOPV [44]. OFETs based on PNDBOPV-DTBT exhibited well-balanced high hole/electron mobilities of 4.68/4.7 cm2 V-1 s-1.
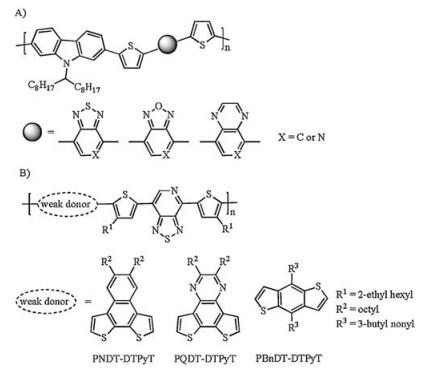
3. D-A conjugated polymers based on pyridal[2, 1, 3]thiadiazoleReplacing the benzene ring in benzo[2, 1, 3]thiadiazole with pyridine was first proposed in the bulk heterojunction (BHJ) polymer solar cells, where the active layer was composed by donor materials and acceptor materials. In order to lower the bandgaps of donor materials to achieve high short-circuit currrent (Jsc), incorporating a more electron-deficient acceptor to construct D-A polymers or small molecules was an effective method. In 2008, Leclerc et al. synthesized several polycarbazole analogues [45] (Fig. 6A), and explored the different performance between benzo[2, 1, 3]thiadiazole and pyridal[2, 1, 3]thiadiazole(PT)-based conjugated polymers. However, the polymerization reactions of pyridine-embedded monomer were hard to achieve high molecular weight, which limited the performance in BHJ solar cells.

|
Download:
|
| Fig. 6. Molecular structures of polycarbazole analogues (A) and pyridal[2, 1, 3]thiadiazole-based conjugated polymers (B). | |
{kind=link}
In 2010, You et al. reported a series of "weak donor-strong acceptor" [46] conjugated polymers based on pyridal[2, 1, 3]thiadiazole. The molecular weight was determined by gel permeation chromatography (GPC) in 1, 2, 4-trichlorobenzene at 135 ℃. These three polymers PNDT-DTPyT, PQDT-DTPyT, and PBnDT-DTPyT (Fig. 6B) showed an Mn of 17.1, 21.7 and 104.4 kg/mol, respectively. Compared with those of benzothiadiazole-based polymers, obviously reduced LUMO levels and slightly decreased HOMO levels were observed by cyclic voltammetry. The optical bandgaps of PNDT-DTPyT, PQDT-DTPyT and PBnDT-DTPyT deduced from UV-vis absorption spectra were 1.53, 1.56 and 1.51 eV, ca. 0.09-0.19 eV smaller than those of benzothiadiazole counterparts. Therefore, this strategy was proved to be effective in designing narrow-bandgap materials.
In order to further lower the bandgap of D-A conjugated polymers based on pyridal[2, 1, 3]thiadiazole, Bazan et al. utilized Lewis acids to coordinate with the nitrogen atoms in pyridal[2, 1, 3]thiadiazole [47]. For example, coordination of the pyridal[2, 1, 3]thiadiazole with electron-deficient boron reagent (such as B(C6F5)3) lowered both HOMO and LUMO levels, which was more pronounced for the LUMO levels.
In addition, Bazan et al. reported the first regioregular PT-based copolymers [48], which exhibited the highest hole mobility of 0.6 cm2 V-1 s-1 due to the high degree of structural order within films. They designed two types of regioregular structures, with the pyridal nitrogen atoms of one type pointed in the same direction, while another type showed the pyridal units alternated in orientation (Fig. 7A). They proposed that the additional dipole moment caused by embedding nitrogen atoms would affect the molecular packing in film. P1-P3 represented the first type, the second type and regiorandom copolymers, respectively. The UV-vis-NIR absorption spectra (Figs. 7B and C) of these polymers' solution at 110 ℃ showed hypsochromic shifts compared with those at room temperature, which resulted from the breakup of aggregated polymer chains. To reveal the impact of regioregularity on the charge mobility, bottom-gate/top-contact OFETs were fabricated through spin-casting. As a result, the hole mobilities of regioregular P1 and P2 were two orders of magnitude higher than that of regiorandom P3. These results revealed the importance of dipole moment on achieving highly ordered molecular orientation.

|
Download:
|
| Fig. 7. (A) Regiochemically precise PT-containing alternating copolymer structures. (B, C) UV-vis-NIR absorption spectra of P1, P2 and P3 (1.0×10-5 g/mL) at 25 ℃ and 110 ℃ in o-DCB. Copied with permission [48]. Copyright 2011, American Chemical Society. | |
{kind=link}
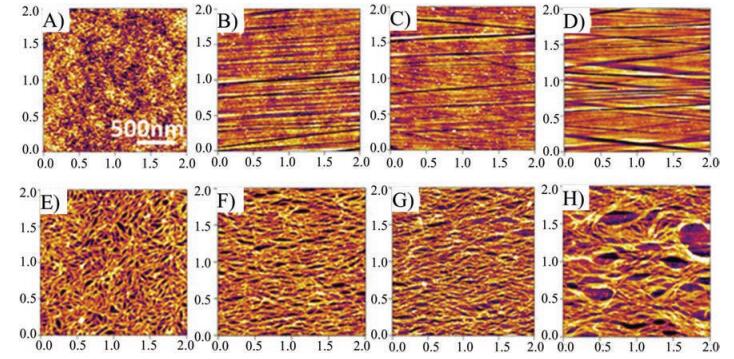
Afterwards, Bazan et al. performed a detailed study on the surface topography of copolymer P2 via atomic force microscopy (AFM) [11, 49]. To achieve high mobility, P2 with high molecular weight (Mn) of 300 kDa and polydispersity (PDI) of 1.5 was synthesized. Figs. 8A-D show the surface morphology of gate dielectrics before and after scratching with diamond nanoparticles. For the untreated dielectric surface, the root-mean-square (RMS) roughness was 0.2 nm. After scratching, the RMS increased to 0.48, 0.49 and 0.86 nm for 100, 250 and 500 nm diamond nanoparticles. In addition, the uniformity was poorer and grooves were observed on the surface. Figs. 8E-H show the morphology of P2 thin films on the unscratched and scratched dielectrics. Long-range orientation and alignment were observed when P2 films were deposited on the nanostructured dielectric substrates. OFETs fabricated on structured dielectric substrates were explored as a function of annealing temperature, and the highest hole mobility of 6.7 cm2 V-1 s-1 was achieved after optimizing several parameters.

|
Download:
|
| Fig. 8. AFM images of dielectric substrate surfaces and related polymer fiber morphology without and with nanostructures: (A) Surface without structures, (B) 100-nm-structured surface, (C) 250-nm-structured surface, (D) 500-nm-structured surface, and (E) polymer fibers on nonstructured surface, (F) polymer fibers on 100-nm-structured surface, (G) polymer fibers on 250-nm-structured surface, (H) polymer fibers on 500-nm-structured surface. Reproduced with permission [11]. Copyright 2012, American Chemical Society. | |
{kind=link}
Subsequently, they proposed a new method [50] to orientate P2 in film by utilizing capillary action. The capillary action contributed to highly oriented crystalline films with a compact lamellar structure, which leads to the hole mobilities of 36.3 cm2 V-1 s-1 for P2 (Mn = 140 kDa) films when the channel length was 140 μm.
In addition, Bazan et al. also explored the relationship between charge mobilities and different mixing ratios of polystyrene (PS) and P2 [51]. They obtained mobilities as high as 2.7 cm2 V-1 s-1 when mixing 90 wt % PS, which was among the highest reported mobilities for majority-insulator blend systems.
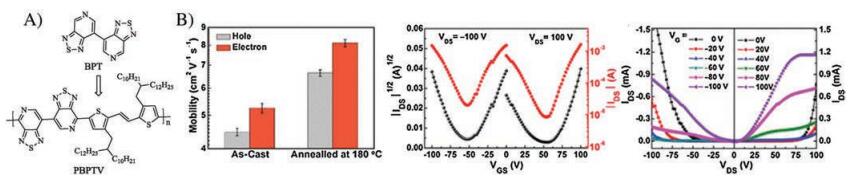
Recently, Liu and coworkers reported the first ambipolar pyridal[2, 1, 3]thiadiazole-based D-A semiconducting polymer (PBPTV) [52], which utilized bispyridal[2, 1, 3]thiadiazole (BPT) as the acceptor unit and alkylated (E)-2-(2-(thiophen-2-yl)vinyl)-thiophene (TVT) as the donor moiety. PBPTV had a molecular weight (Mn) of 22.7 kDa and narrow PDI of 2.23. Cyclic voltammetry measurements revealed its HOMO level of -5.61 eV and LUMO level of -3.66 eV, which are suitable for ambipolar charge transport. Top-gate/bottom-contact OFET devices were fabricated, and the hole and electron mobilities of 4.47 and 5.25 cm2 V-1 s-1 were obtained respectively for the as-cast thin films (Fig. 9). Further annealing of the thin films gave the best performance with the average hole and electron mobilities of 6.64 and 8.13 cm2 V-1 s-1. AFM images of PBPTV thin films showed small surface roughness. Moreover, 2D grazing-incidence wide-angle X-ray scattering (GIWAXS) indicated that the polymer chains arranged mainly in an edge-on orientation and the annealing treatment led to higher degree ordering, which is beneficial to the carrier transport.

|
Download:
|
| Fig. 9. (A) Molecular structures of BPT acceptor and its copolymer PBPTV; (B) Transfer and output curves of the hero FET device. Copied with permission [52]. Copyright 2017, American Chemical Society. | |
{kind=link}
4. Summary and perspective
In this review, we outlined several design strategies and recent progress in pyridine-embedded D-A conjugated polymers. Based on these results, we can conclude that introducing sp2-nitrogen atoms into polymer backbones has several advantages: ⅰ) lowering the LUMO levels to realize the ideal charge injection; ⅱ) enhancing the coplanarity via noncovalent interactions to lock the conformation; ⅲ) providing coordination sites with Lewis acids to lower the energy levels, especially the LUMO levels; ⅳ) producing dipole moment to achieve ordered molecular orientation. However, some challenges still exist, such as designing new types of pyridine-containing D-A conjugated polymers, better controlling the molecular packing, and substantially improving the device performance. Therefore, effort on molecular design and device processing engineering is still needed to apply this strategy in organic electronics.
AcknowledgmentsThis work was supported by National Key R & D Program of China (No. 2017YFA0204701), National Natural Science Foundation of China (Nos. 21722201, 21790360, 21420102005), and the Major State Basic Research Development Program (No. 2015CB856505) from the MOST.
| [1] |
S. Rochat, T.M. Swager, J. Am. Chem. Soc. 135 (2013) 17703-17706. DOI:10.1021/ja4095395 |
| [2] |
D. Izuhara, T.M. Swager, J. Am. Chem. Soc. 131 (2009) 17724-17725. DOI:10.1021/ja906513u |
| [3] |
P.M. Beaujuge, J.M. Frechet, J. Am. Chem. Soc. 133 (2011) 20009-20029. DOI:10.1021/ja2073643 |
| [4] |
J. Mei, Y. Diao, A.L. Appleton, L. Fang, Z. Bao, J. Am. Chem. Soc. 135 (2013) 6724-6746. DOI:10.1021/ja400881n |
| [5] |
A.J. Heeger, Chem. Soc. Rev. 39 (2010) 2354-2371. DOI:10.1039/b914956m |
| [6] |
T. Lei, Y. Cao, Y. Fan, et al., J. Am. Chem. Soc. 133 (2011) 6099-6101. DOI:10.1021/ja111066r |
| [7] |
J. Mei, D.H. Kim, A.L. Ayzner, M.F. Toney, Z. Bao, J. Am. Chem. Soc. 133 (2011) 20130-20133. DOI:10.1021/ja209328m |
| [8] |
R. Stalder, S.R. Puniredd, M.R. Hansen, et al., Chem. Mater. 28 (2016) 1286-1297. DOI:10.1021/acs.chemmater.5b03968 |
| [9] |
J. Li, Y. Zhao, H.S. Tan, et al., Sci. Rep.-UK 2 (2012) 754. DOI:10.1038/srep00754 |
| [10] |
C.B. Nielsen, M. Turbiez, I. McCulloch, Adv. Mater. 25 (2013) 1859-1880. DOI:10.1002/adma.v25.13 |
| [11] |
H.R. Tseng, L. Ying, B.B. Hsu, et al., Nano Lett. 12 (2012) 6353-6357. DOI:10.1021/nl303612z |
| [12] |
S. Wang, M. Kappl, I. Liebewirth, et al., Adv. Mater. 24 (2012) 417-420. DOI:10.1002/adma.201103057 |
| [13] |
C. Wang, H. Dong, W. Hu, Y. Liu, D. Zhu, et al., Chem. Rev. 112 (2012) 2208-2267. DOI:10.1021/cr100380z |
| [14] |
H. Yan, Z. Chen, Y. Zheng, et al., Nature 457 (2009) 679-686. DOI:10.1038/nature07727 |
| [15] |
H. Huang, Z. Chen, R. PonceOrtiz, et al., J.Am.Chem.Soc. 134 (2012) 10966-10973. DOI:10.1021/ja303401s |
| [16] |
T. Lei, X. Xia, J.Y. Wang, C.J. Liu, J. Pei, et al., J. Am. Chem. Soc. 136 (2014) 2135-2141. DOI:10.1021/ja412533d |
| [17] |
J.H. Dou, Y.Q. Zheng, Z.F. Yao, et al., Adv. Mater. 27 (2015) 8051-8055. DOI:10.1002/adma.201503803 |
| [18] |
T. Lei, J.H. Dou, Z.J. Ma, et al., Chem. Sci. 4 (2013) 2447-2452. DOI:10.1039/c3sc50245g |
| [19] |
Z. Liang, Q. Tang, J. Xu, Q. Miao, Adv. Mater. 23 (2011) 1535-1539. DOI:10.1002/adma.201004325 |
| [20] |
U.H. Bunz, J.U. Engelhart, B.D. Lindner, M. Schaffroth, Angew. Chem. Int. Ed. Engl. 52 (2013) 3810-3821. DOI:10.1002/anie.v52.14 |
| [21] |
S. Yang, D. Liu, X. Xu, Q. Miao, Chem. Commun. 51 (2015) 4275-4278. DOI:10.1039/C5CC00537J |
| [22] |
U.H. Bunz, Acc. Chem. Res. 48 (2015) 1676-1686. DOI:10.1021/acs.accounts.5b00118 |
| [23] |
L. Ji, A. Friedrich, I. Krummenacher, et al., J. Am. Chem. Soc. 139 (2017) 15968-15976. DOI:10.1021/jacs.7b09460 |
| [24] |
V. Lami, D. Leibold, P. Fassl, et al., Solar RRL 1 (2017) 1700053. DOI:10.1002/solr.201700053 |
| [25] |
H. Huang, L. Yang, A. Facchetti, T.J. Marks, et al., Chem. Rev. 117 (2017) 10291-10318. DOI:10.1021/acs.chemrev.7b00084 |
| [26] |
Z.F. Yao, J.Y. Wang, J. Pei, Cryst. Growth Des. 18 (2018) 7-15. DOI:10.1021/acs.cgd.7b01385 |
| [27] |
J. Mei, K.R. Graham, R. Stalder, J.R. Reynolds, Org. Lett. 12 (2010) 660-663. DOI:10.1021/ol902512x |
| [28] |
T. Lei, J.H. Dou, J. Pei, Adv. Mater. 24 (2012) 6457-6461. DOI:10.1002/adma.v24.48 |
| [29] |
J. Huang, Z. Mao, Z. Chen, et al., Chem. Mater. 28 (2016) 2209-2218. DOI:10.1021/acs.chemmater.6b00154 |
| [30] |
Y. Lu, Y. Ding, J.Y. Wang, J. Pei, Chin. J. Org. Chem. 36 (2016) 2272-2285. DOI:10.6023/cjoc201606015 |
| [31] |
R.S. Ashraf, A.J. Kronemeijer, D.I. James, H. Sirringhaus, I. McCulloch, Chem. Commun. 48 (2012) 3939-3941. DOI:10.1039/c2cc30169e |
| [32] |
N. Zhao, N. Ai, M. Cai, et al., Polym. Chem.-UK 7 (2016) 235-243. DOI:10.1039/C5PY01488C |
| [33] |
G.W.P. Van Pruissen, F. Gholamrezaie, M.M. Wienk, R.A.J. Janssen, J. Mater. Chem. 22 (2012) 20387-20393. DOI:10.1039/c2jm34668k |
| [34] |
T. Lei, J.Y. Wang, J. Pei, Chem. Mater. 26 (2014) 594-603. DOI:10.1021/cm4018776 |
| [35] |
M. Cai, X. Bao, Y.F. Liu, et al., Chem. Mater. 29 (2017) 9162-9170. DOI:10.1021/acs.chemmater.7b03025 |
| [36] |
L. Xu, Z. Zhao, M. Xiao, et al., ACS Appl. Mat. Interfaces 9 (2017) 40549-40555. DOI:10.1021/acsami.7b13570 |
| [37] |
F. Bouchikhi, F. Anizon, P. Moreau, Eur. J. Med. Chem. 43 (2008) 755-762. DOI:10.1016/j.ejmech.2007.05.012 |
| [38] |
R. Sriram, C.N. Sesha Sai Pavan Kumar, N. Raghunandan, et al., Synth. Commun. 42 (2011) 3419-3428. |
| [39] |
G. de Miguel, L. Camacho, E.M. Garcia-Frutos, J. Mater. Chem. C 4 (2016) 1208-1214. |
| [40] |
Y. Lu, Y. Liu, Y.Z. Dai, et al., Chem.-Asian J. 12 (2017) 302-307. DOI:10.1002/asia.v12.3 |
| [41] |
Y.Z. Dai, N. Ai, Y. Lu, et al., Chem. Sci. 7 (2016) 5753-5757. DOI:10.1039/C6SC01380E |
| [42] |
G. Zhang, Y. Dai, K. Song, et al., Polym. Chem.-UK. 8 (2017) 2381-2389. DOI:10.1039/C7PY00295E |
| [43] |
T. Lei, J.H. Dou, X.Y. Cao, J.Y. Wang, J. Pei, Adv. Mater. 25 (2013) 6589-6593. DOI:10.1002/adma.201302278 |
| [44] |
K. Shi, W. Zhang, D. Gao, et al., Adv. Mater. 30 (2018) 1705286. DOI:10.1002/adma.201705286 |
| [45] |
N. Blouin, A. Michaud, D. Gendron, et al., J. Am. Chem. Soc. 130 (2008) 732-742. DOI:10.1021/ja0771989 |
| [46] |
H. Zhou, L. Yang, S. Stoneking, W. You, ACS Appl. Mater. Interfaces 2 (2010) 1377-1383. DOI:10.1021/am1000344 |
| [47] |
G.C. Welch, G.C. Bazan, J. Am. Chem. Soc. 133 (2011) 4632-4644. DOI:10.1021/ja110968m |
| [48] |
L. Ying, B.B. Hsu, H. Zhan, et al., J. Am. Chem. Soc. 133 (2011) 18538-18541. DOI:10.1021/ja207543g |
| [49] |
Z.B. Henson, G.C. Welch, T. van der Poll, G.C. Bazan, J. Am. Chem. Soc. 134 (2012) 3766-3779. DOI:10.1021/ja209331y |
| [50] |
C. Luo, A.K. Kyaw, L.A. Perez, et al., Nano Lett. 14 (2014) 2764-2771. DOI:10.1021/nl500758w |
| [51] |
M.J. Ford, M. Wang, S.N. Patel, et al., Chem. Mater. 28 (2016) 1256-1260. DOI:10.1021/acs.chemmater.5b04774 |
| [52] |
C. Zhu, Z. Zhao, H. Chen, et al., J. Am. Chem. Soc. 139 (2017) 17735-17738. DOI:10.1021/jacs.7b10256 |