 2019, Vol. 30
2019, Vol. 30 


b School of Chemistry and Chemical Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
The transformation of biomass into chemicals has a long history. At the beginning of the 20th century, a large number of industrial products were manufactured from wood-based resources and crops [1]. During the past 100 years, the interest on utilization of bio-based resources is inversely correlated with the oil supply and price [2]. However, petroleum resources are limited. Developing inexhaustible renewable natural resource as the alternative is urgently desired in the long term, which is the necessary requirement for sustainable development [2, 3].
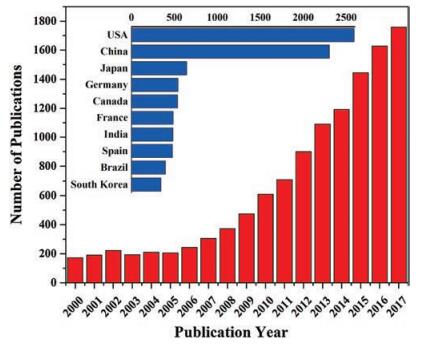
As one of the most abundant renewable carbon resource on earth, lignocellulosic biomass attracts most of the interest [4]. Lignocellulose is mainly composed of the semi-crystalline polysaccharide cellulose, amorphous multicomponent polysaccharide hemicelluloses, and amorphous aromatic polymer lignin [5, 6]. Lignin in plant fills in the gaps between cellulose and hemicelluloses, which acts like adhesives that keeps the lignocellulose matrix together and adds strength and rigidity to cell walls [7, 8]. The lignin content depends on the plant taxonomy, about 30% in conifer wood, 20%-25% in broadleaf wood and 10%-15% in grass, respectively [6, 9-11]. A number of technologies have been developed for lignin fractionation from lignocellulosic biomass [12, 13], and pulp industry yields large amount of kraft lignin annually. Among the three components of lignocellulose, lignin is the most complex, and its utilization for value-added chemicals is a severer challenge compared to cellulose and hemicelluloses [14]. But great attentions should be paid to lignin, because lignin is the only abundant renewable natural aromatic biopolymer, which can be a sustainable candidate feedstock for aromatic chemicals [6, 15]. Although some aliphatic and cycloaliphatic compounds are industrially available from cellulose [16], starch [16], or triglycerides [17], many key chemicals are aromatic compounds and ultimately derived from petroleum [18]. However, currently almost 98 wt% of lignin as waste stream is combusted to produce heat and power for the biorefinery in the pulp and paper industry [19, 20]. It is not only harmful to the environment but also a waste of resource [21]. The rest lignin is used in low value-commercial applications, such as a low-grade fuel for heat and power [22], water reducing dispersing [23], emulsifying agents [24], phenolic resin [25] and polyurethane foams [26]. The potential of lignin is far from being fully developed. The good news is that more and more researchers are recognizing the great value of lignin, and researches on lignin conversion into platform chemicals and alternative fuels are booming in the last decade (Fig. 1).

|
Download:
|
| Fig. 1. Publications on lignin transformation from the beginning of this century (derived from Web of Science using "lignin utilization/transformation/conversion/depolymerization" as the key words). | |
The basic premise for lignin utilization is to depolymerize it into small molecules, and there have been several systematic reviews [6, 27, 28]. The lignin deconstruction strategies can be broadly classified into acidolysis [29-32], base depolymerization [33-36], pyrolysis [37-40], gasification [41-43], oxidation depolymerization [45-47], hydrotreating (including hydrogenation [45-47], hydrodeoxygenation [48-50] and hydrogenolysis [51-56]) and the combined processes [57-60]. Based on the types of reactions involved, the above strategies can be simplified into three modes: (1) oxidizing reaction (nitrobenzene, metal oxides, O2, air, H2O2, or other peralcohol was used as an oxidant), (2) reducing reaction (H2, hydride or hydrogen donor solvent was used a reductant), (3) non-redox reaction. Oxidation reaction generally occurs at relatively low temperatures (below 250 ℃) [6, 28]. In the depolymerization system, oxidation reaction involves the cleavage of the aryl ether bonds (β-O-4, 4-O-5, α-O-4), C-C bonds (β-5, β-β, β-1 or 5-5), or other linkages in the lignin. Meanwhile oxidation of the functional groups (hydroxyl, aromatic ring or others) in lignin also occurs and produce polyfunctional aromatic compounds. It favors the production of various target fine chemicals, such as aromatic alcohols, aldehydesand acids. However, the oxidation process cannot be easily controlled, and the alcohols, aldehydes and acids were usually formed simultaneously in one-pot, which increased the cost of products separation in industry. Reducing reaction involves thermal hydroprocessing reductions of lignin in hydrogen atmosphere at high temperatures ranging from 100 ℃ to 350 ℃ [6, 61, 62], including hydrogenolysis, hydroalkylation, hydrodeoxygenation, hydrogenation, and integrated hydrogen-related reactions [6]. In reducing depolymerization system, not only the cleavage of ether linkages in lignin occurs, but also the functional groups (hydroxyl, aldehyde and carboxyl group, carbon-carbon double bonds and the aromatic rings) in lignin can be reduced. But compared to oxidizing reaction, the selectivity of these reactions is easier to be controlled, which is benefit for highly selective target fine chemicals. It is by far one of the most popular and efficient technologies applied in depolymerization of lignin into oligomers, phenolsand further upgrading productions such as aromatic and aliphatic hydrocarbons. Non-redox reaction mainly contains acidolysis, base depolymerization, pyrolysis and gasification. Both acid (typically at 0-200 ℃) and base (100-300 ℃) can cleavage the ether linkage and C-C bond in lignin into low depolymerized lignin and monomeric phenols. Acid environment is liable to condensation reaction of the lignin fragments, which suppress further depolymerization of lignin, while base can efficiently break ether linkage and C-C bond to form various depolymerized productions. Pyrolysis (typically at 450-700 ℃) is a process that directly produces bio-oil from lignocellulosic biomass. Gasification involves two different commercial processes: Fischer-Tropsch synthesis or methanol/dimethyl ether synthesis to produce syngas (hydrogen and carbon monoxide) from a range of real lignin feedstocks.
Although much effort has been made on transformation of lignin into value-added chemicals, almost all of the current technologies provide a mixture of numerous products, such as oligomers, phenols, aldehydes, ketones, acids, esters, etc. Isolating each of these compounds is currently not possible and even less economically viable [18]. Even if harvesting the molecules of interest, the separation and purification process can also be extremely energy intensive, and sometimes it is challenging [63]. For example, the separation of vanillin from lignin oxidation mixture is rather complex, which can generate huge amount of caustic soda wastes (160 kg/kg vanillin) [64]! Separation of vanillin and syringaldehydefrom the lignin oxidation products was a subject of intense researches, and the separation problem prevent the preparation of syringaldehyde from lignin for decades [65]. The problem of complex mixed products may be one of the main limiting factor for the scaling high-value utilization of lignin in industry. The realization of lignin industrialization presupposes not only high conversion of lignin, but also high selectivity of depolymerization products, which can reduce energy consumption and cost. Thus, it is desperately desired to develop highly selective approaches to lignin transformation and avoid the formation of side products as less as possible at the very beginning. Some researchers have been aware of the significance of "selectivity", and great efforts are being made to harvest product with high selectivity from lignin. But selective production of target fine chemicals or platform chemicals from lignin is a severe challenge because of the complex structure. Up to now, there is only one report that claimed to achieve one single product from lignin [66]. The term "selective" in other literatures about lignin utilization actually means that a specific class of chemicals sharing the same functional groups are obtained. Here in this minireview, we follow this convention. Although there have been numerous comprehensive reviews on lignin valorization, selective transformation of lignin has not been focused. Thus, we write this minireview to highlight this key issue.
The thesis of this review focused on the selective transformation of lignin into value-added chemicals. Contributions from model compounds are not included, considering the selectivity differs a lot from real lignin due to its much more complicated structures. For the researches on model molecules, the readers are suggested to consult other reviews [30, 67]. In the first section, we give a brief introduction to the structure of lignin. Then in the second section, we summarized typical researches focused on selectivity utilization of lignin to produce target chemicals, organized by the types of products (phenols, phenolic aldehydes, carboxylic acids, alkanes, arenes and other compounds). Finally, we make a brief discussion on the challenges and opportunities for selective transformation of lignin.
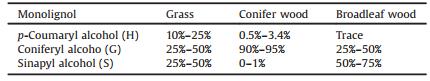
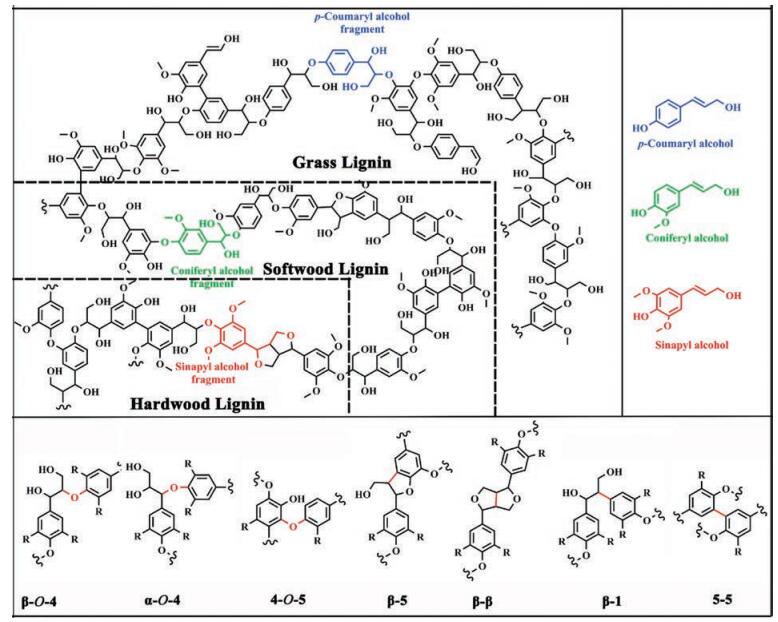
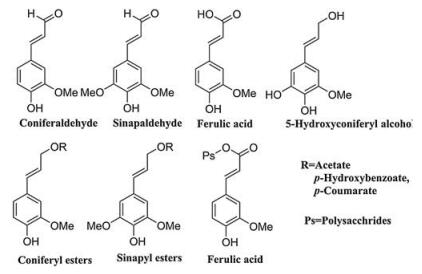
2. Structure of ligninIn nature, lignin presents a complex three-dimensional amorphous structure, arising from enzymatic dehydrogenative polymerization of three different cinnamyl alcohol monomers: sinapyl alcohol (syringyl (S) units), coniferyl alcohol (guaiacyl (G) units) and p-coumaryl alcohol (p-hydroxyphenyl (H) units) [68], as shown in Fig. 2. The content and the ratio of monomers in lignin varies in different types of plants [10, 69]. Generally, lignin from conifer wood has more guaiacyl (G) units than the other two monolignols, broadleaf wood only contents guaiacyl (G) and syringyl (S) units, and grass lignin presents a mixture of all three monolignols [11]. Table 1 shows the typical content of the three primary lignin units in different plants. Based on composition and abundance, lignin can be classified into three categories: G-lignin, G-S-lignin and G-S-H-lignin [6, 9]. However, with the going deep of the research work, researchers found that some other aromatic species also can involve in the formation of the lignin, which includes coniferaldehyde, sinapaldehyde, ferulic acid, 5-hydroxyconiferyl alcohol and the above monolignols containing acylated groups (acetate, p-hydroxybenzoate, or p-coumarate) [70-72], as shown in Fig. 3. One of the most significant obstacle of high-value utilization of lignin is due to its heterogeneity of structure, which depends on the biomass sources, wood type and even some growing parameters [73].

|
Download:
|
| Fig. 2. Structure of lignin and the main inter-subunit linkages present in lignin. | |
|
|
Table 1 Abundance of the three basic lignin units in different types of plants [6]. |

|
Download:
|
| Fig. 3. Other lignin precursors found in lignin structure [68]. | |
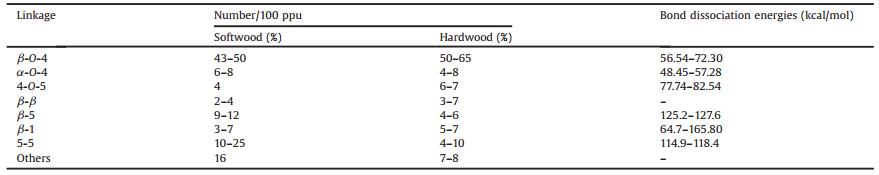
These building units of lignin are predominantly bonded by a series of C-O and C-C linkages, such as β-O-4 (β-aryl ether), 4-O-5 (diaryl ether), α-O-4 (α-aryl ether), β-5 (phenylcoumaran), β-β (resinol), β-1 (spirodienone) or 5-5, to form a very complex polymer [28, 62]. Fig. 2 presents the representative structures of these linkages in lignin, and Table 1 lists the typical content of the major linkages in different types of plants. Although the contents of these linkages are related to the type of lignocellulosic biomass, generally more than 50% of the linkages in lignin are ether linkages. Importantly, among these ether linkages, the β-O-4 (β-aryl ether) linkage is dominant in both softwood and hardwood, representing from ca. 43% (G-lignin) to ca. 65% (G-S-lignin) as indicated in Table 2 [74]. Moreover, it is reported that proportion of resistant linkages (β-β, β-5, β-1 and 5-5) in softwood lignin is higher than that in hardwood lignin, because the C5 positions of guaiacyl (G) units in softwood lignin tends to commit coupling reaction to form resistant linkages whereas those in syringyl (S) unit is sterically inhibited due to addition of methoxy group [6, 74]. Therefore, G-type lignin (softwood lignin) contains much higher condensation degree than hardwood lignin. Besides, due to steric effect, the additional methoxy groups in syringyl (S) unit could inhibit formation of other linkages, leading to more linear structures in the hardwood lignin compared with softwood lignin. Grass lignin involves other noncanonical monolignols (Fig. 3) in its biosynthesis process and it special in structure [75]. The representative model structure of lignin is illustrated in Fig. 2. It should be noted that these model structures of lignin are not the real structure in raw lignocellulosic biomass, but it can represent the linkages between adjacent building units, as well as the contents in lignin.
|
|
Table 2 Content distribution and bond dissociation energies of the lignin linkages in hardwood and softwood [6, 74, 75, 76, 77]. |
{kind=link}
{kind=link}
{kind=link}
3. Selective transformation of lignin
The basis of lignin transformation is the cleavage of the linkages (intermolecular C-O and C-C bonds) between the monomers. As shown in Table 1, the bond energies of C-O bonds are much lower than those of the C-C bonds. Thus, C-O bond is considered as cleavable bonds, while C-C bonds are non-cleavable linkage [78]. Fortunately, the cleavable C-O bonds, especially β-O-4 type, are the major linkage in lignin (Table 2), which indicates that lignin can be depolymerized into small molecules under appropriate conditions. Numerous methods have been proposed for the depolymerization of lignin, and various products can be obtained, heavily depending on the treatment methods, reaction conditions, and lignin source [28]. Phenols, aldehydes, carboxylic acids, alkanesand alkenes can be derived from lignin with high selectivity.
3.1. PhenolsPhenols were important platform chemicals with applications in many fields, such as biofuel [79-81] and key precursors of plastics/cosmetics/pharmaceutics [82]. Substituted phenols are the most commonly reported derivates from lignin. Numerous methods have been developed for the selective transform of lignin into phenols.
3.1.1. HydrogenolysisHydrogenolysis is one of the most employed methods to cleavage ether bonds [83-88]. The most widely used catalyst are supported metal (Pt, Ru, Pd, Rh) particles or metal organic complex [83]. Although the cleavage of different types of ether bonds can be well controlled in model compounds, there's only a few reports on hydrogenolysis of lignin into phenols with high selectivity. For example, Wang, Zhang and co-workers achieved the selectively hydrogenolysis of Kraft lignin into phenols over Pd/C using choline-derived ionic liquids as the reaction media [82]. Under optimum reaction condition, the conversion of Kraft lignin and the selectivity to phenol (PL) and catechol (COL) reached 20.3%, 18.4%, and 18.1%, respectively [82]. One of the challenges to obtain phenols from lignin by hydrogenolysis is that the aromatic ring can also be hydrogenated. Although noble metals have demonstrated efficient catalytic hydrogenation activity, it is sometimes difficult to achieve high selectivity. As alternatives, non-noble metal catalysts are attracting researchers interest for they are easier to control the selectivity. For example, the hydrogenolysis product of lignin over Ni-based catalyst can be significantly tuned by the solvent. Rinaldi et al. found that the Lewis basicity of the solvent is an important parameter in the reaction performance with Raney Ni [89]. Basic solvents can prevent the hydrogenation of the aromatic ring while has no effect on the hydrogenolysis of the ether bond, resulting in high selectivity to phenol products. The hydrogenolysis of lignin in methanol is found to produce mostly phenols over Raney Ni. Cu-doped porous metal oxides were also found to be efficient for promoting the selective hydrogenolysis of lignin [52]. Temperature affected the products significantly, and C9 catechols were obtained with high selectivity at 140-220 ℃. Especially, 4-(3-hydroxypropyl)-catechol was the major product and could be isolated in high purity at 140 ℃.
A commonly existed problem in lignin depolymerization is that the depolymerized fragments can also condense to form non-cleavable C-C bonds and form insoluble materials [90], which hampers the conversion of lignin into well-defined aromatic chemicals [30]. Luterbacher et al. put forward an solution to facilitate lignin monomer production by pretreating the lignin with formaldehyde, which prevented lignin condensation by forming 1, 3-dioxane structures with lignin side-chain hydroxyl groups [51]. Via this pretreatment, the soluble lignin fraction can be converted to guaiacyl and syringyl monomers at near theoretical yields during subsequent hydrogenolysis. The yield of the monomers reached up to 47 mol% and 78 mol% for Klason lignin of beech and high-syringyl transgenic poplar, respectively.
As lignin is symbiotic with polysaccharide (cellulose and hemicelluloses) in nature, researchers also make efforts to directly utilize lignin using lignocellulose or even unpretreated wood as starting material [78, 91-93]. This alternative strategy is known as lignin-first approach [94], which is considered to have the potential to yield nearly theoretical amounts of phenolic monomers by performing solvolytic delignification and lignin depolymerization [91]. For example, Sels et al. presented a catalytic lignocellulose biorefinery process, where birchsawdust is efficiently delignified through simultaneous solvolysis and catalytic hydrogenolysis Ru/C in methanol [95]. The yield of phenolic monomers was above 50% in the lignin oil, mainly 4-n-propylguaiacol and 4-n-propylsyringol. Hensen and co-workers reported an approach to efficently release lignin fragements from lignocellulose by lewis acid metal triflates. Combined with metal-catalyzed (Pd/C) hydrogenolysis, the lignin in lignocellulose can be converted to C9 phenol derivates in 46 wt% yield, leaving cellulose largely untouched [94]. Ni-based catalysts are also widely used in the lignin-first strategy. Abu-Omar and co-workers reported the catalytic conversion of lignin in Miscanthus over Ni/C catalyst. By modification of the reaction conditions (temperature and pressure of H2), saturated or unsaturated branched products can be obtained selectively [96]. Optimal conditions gave 69% yield of phenol derivates. Zhang et al. developed a carbon supported Ni-W2C catalyst which can directly catalyze the hydrogenolysis of raw woody biomass into two groups of chemicals [55]. The reaction was conducted at 235 ℃ and 6 MPa H2 for 4 h in water media. Cellulose and hemicelluloses were converted to ethylene glycol and other diols, while lignin can be selectively converted into phenols with a total yield of 46.5% (mainly guaiacylpropanol, syringylpropanol, guaiacylpropane and syringylpropane). Recently, Sels et al. investigated the reductive catalytic fractionation of wood over Ni/Al2O3, producing a lignin oil that contains over 40% phenolic monomers, of which 70% consists of 4-n-propanolguaiacol and −syringol [91]. The role of the Ni/Al2O3 catalyst was also well addressed. Bimetal catalysts were also employed, for example, Ni-Fe alloy catalyst supported on activated carbon was also used for hydrogenolysis of lignin, which acheived 23.2 wt% total yield of monomers at 225 ℃ under 2 MPa H2, mainly propylguaiacol and propylsyringol.
3.1.2. AcidolysisAcidolysis is one of the most prevalent methods for the fractionation of lignocellulose into its main components [28]. Both Bronsted acid and Lewis acid can cleavage the C-O bond in lignin. For example, Stahl et al. proposed a method for the depolymerization of oxidized lignin under mild conditions (110 ℃) in aqueous formic acid [31]. More than 52% of the original oxidized lignin was converted to well-defined phenol derivatives, among which syringyl and guaiacyl-derived diketones are two of the major depolymerization products (19.8%). However, this method is not as efficient for normal unoxidized lignin, and the yield of low-molecular-mass aromatics were only 7.2 wt%. Catalytic amount of appropriate Lewis acid can be effective for lignin depolymerization. Metal triflates is found to be an excellent promoter. For example, Bruijnincx and co-workers reported a route to depolymerize lignin by a combination of Lewis acid metal triflates and rhodium complexes. The selectivity towards either 4-methylphenols or 4-(1-propenyl)phenols can be controlled by varying the amount and strength of the Lewis acid catalyst [29]. Barta and co-workers also proved that Fe(OTf)3 can effectively catalyze the conversion of lignin into phenolic C2-acetal products [30, 97].
The recondensation phenomenon is also severe in acidolysis process. Barta and co-workers revealed the causes of this problem is that the aldehyde intermediates are very reactive, and easy to form C-C bond with other species [98]. They proposed a strategy to increase the yield of aromatic monomers by in situ conversion of reactive intermediates in the acid-catalyzed depolymerization of lignin [98]. Three specific methods were developed, that is, in situ catalytic hydrogenation, in situ catalytic decarbonylation and in situ conversion of the aldehyde intermediates to acetals. The efficiency and mechanism was verified by model compounds, and the methods were demonstrated to be viable for real lignin [98].
3.1.3. Non-hydrogen reductive depolymerizationStrictly, hydrogenolysis also belongs to reductive depolymerization. Since it is extremely important, we have discussed it independently, and here in this section we just review selective reductive depolymerization of lignin using non-hydrogen reductant. It has been demonstrated in previous paragraphs that Ni-based catalyst is efficient and selective for hydrogenolysis of ether bonds [83, 85]. Actually, it can also promote the non-hydrogen reductive depolymerization of lignin and has been widely used. For example, Luque and Labid proposed an approach to reductive depolymerize organosolv lignin over Ni/AlSBA-15 catalyst using formic acid as the reductive reagent as well as the solvent under microwave iradation. It yielded simple phenolic monomeric products, such as desaspidinol, syringaldehyde, and syringol [99]. Alcohols are frequently-used as solvent in lignin treatment, and they can act as hydrogen source at appropriate reaction condition. For example, Wang and Xu et al. showed that Ni/C catalyst can selectively promote the transformation of lignin into monomeric phenols, propylguaiacol and propylsyringol [54]. The seletivity of the phenol products can reach up to more than 90% and 50% converison of birch wood lignin. Alcohols, such as methanol, ethanol and ethylene glycol, are suitable solvents for the depolymerization, and it was demonstrated that alcohol played dual role in the cleavage of the linkage of lignin. It acted as the nucleophilic reagent for C-O-C cleavage in alcoholysis reaction and functioned as hydrogen source in the reduction cleavage steps [54]. The reaction mechanism was shown in Fig. 4. Besides organic reductant, metal Zn is also used for selectively reductive depolymerization of lignin. Westwood et al. developed a one-pot method which involves an initial catalytic oxidation (using DDQ and O2) of the β-O-4 linkages in lignin followed by zinc-mediated cleavage of the C-OAryl bond [100]. Application of this depolymerization method to birch lignin followed by extraction and purification gave isolated phenolic monomers in 6 wt% yield, among which 80% is 3-hydroxy-1-(4-hydroxy-3, 5-dimethoxyphenyl)propan-1-one.

|
Download:
|
| Fig. 4. Mechanism of the formation of monomeric phenols by hydrotreating lignin using alcohols as hydrogen source over Ni/C catalyst [54]. Adapted with permission [54]. Copyright 2013, Royal Society of Chemistry. | |
{kind=link}
In general, harsh reaction conditions (high temperature, high pressure, strong acid or base) are required for the cleavage of lignin, which lead to problematic condensation of the depolymerized fragment via C-C bond formation [101]. In 2015, Cantat and co-workers reported a route to reductive depolymerization of lignin under ambient conditions using hydrosilanes as reductants and B(C6F5)3 as a Lewis acid catalyst [101]. This is the first example of metal-free catalysis for the reductive depolymerization of lignin. Different lignin preparations derived from 15 gymnosperms and angiosperms species were successfully depolymerized using this method, which yielded a narrow distribution of phenol derivatives. The isolated yiled of the phenol monomers (e.g., 5-(3-hydroxypropyl)benzene-1, 2, 3-triol and 4-propylbenzene-1, 2-diol) were 7 wt%-24 wt% from lignin and 0.5wt%-2.4 wt% from wood. Interestingly, by tuning the catalytic conditions and selecting the wood source and the lignin extraction method, several different aromatic products can be isolated selectively from wood [101]. Together with the work of Westwood's, this two contributions are the rare examples that lignin is depolymerized under mild conditions. However, the reagent in the reactions are a bit expensive, more economically viable approach are yet to be developed.
3.1.4. Other methodsOther methods such as thermolysis and solvolysis are also developed for selective transformation of lignin into phenols. For example, Li and Hu et al. achieved the selective conversion of lignin in corncob residue to monophenols via a two-step process without significant degradation of cellulose [102]. Lignin was thermotreated in THF at 300 ℃, and the yield of total monophenols reached up to 24.3 wt%, among which 86.8% were the predominant products, 4-ethylphenol(10.5 wt%), 2, 6-dimethoxyphenol (6.6 wt%), and 4-ethylguaiacol (4.0 wt%). The authors also treated lignin in Phyllostachys heterocycla cv. pubescens in an ethanol solvothermal system at 220 ℃, and 45.3% of the lignin was degraded at 2 h. A maximum yield of 10.6 wt% of 4-ethyl phenols was obtained under optimized conditions [103]. Subsequently, they extended the solvent to Na2CO3-H2O-THF solution, and the total yield of monophenols reached up to 26.9 wt%. Therein, the reaction mechanism of the cleavage of inter- and intramolecular linkages was investigated in details [79]. Chornet and co-workers conducted a hydrolytic based-catalyzed depolymerization of a steam exploded aspen lignin, and the yield of the monomers are 28 wt%, among which 89.4% is phenol derivates [63]. The authors also investigated the separation methods of these monomers.
Oxidation is also an important method for ether bond cleavage [44, 104, 105]. However, due to over-oxidation, it is challenging to get phenol derivates with high selectivity. For example, Zhang et al. reported a highly efficient lignin-to-monomeric phenolic compounds conversion method based on peracetic acid (PAA) treatment [106]. Diluted acid pretreated corn stover lignin and steam exploded spruce lignin were treated with this method, and the highest yield of phenolic compounds reached up to 47%. However, the "phenolic compounds" contains quantities of acid products, such as p-hydroxybenzoic acid, vanillic acid, syringic acid and 3, 4-dihydroxybenzoic acid. Nevertheless, selective oxidation of the β-O-4 linkages in lignin has been demonstrated to be feasible [107], which weakens the C-O and C-C bond energy [104]. The resulted oxidized lignin is easier to be depolymerized with other method. Thus, a two-step oxidation depolymerization process is an efficient approach to the selective transformation of lignin, such as the work of Stahl's group [31] and Westwoods' group [100]which has been reviewed in previous paragraphs.
3.2. Phenolic aldehydesPhenolic aldehydes are also frequently reported as major depolymerization products of lignin, which are mainly derived from p-hydroxyphenyl (H), guaiacyl (G), or syringyl (S) units [18].
The most important phenolic aldehyde derived from lignin is vanillin, which is currently one of the only molecular phenolic compounds manufactured on an industrial scale from biomass [18]. The lignin-to-vanillin process can be traced back to the year of 1936 [18], and nowadays 15% of the overall vanillin are produced from lignin [64]. However, only lignin from the sulfite pulping process is used to prepare vanillin industrially [18], and the price of lignin-based vanillin is consistently ten times higher than that of guaiacol-based vanillin [109]. Recent years, lignin-to-vanillin researches focus on extending the feedstock to all kinds of lignin, improving the selectivity and understanding the corresponding mechanism [64, 110]. For more details about vanillin production from lignin, please refer to the corresponding reviews [18, 65].
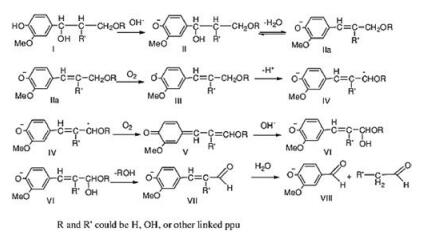
Lignin is not naturally rich of carbonyl groups in raw lignocellulose. The carbonyl groups mainly come from the oxidation of the hydroxyls [67, 112] or the rearrangement of the enolic structures derived from hydrolysis [110]. Thus, oxidation depolymerization and hydrolysis are the main methods to obtain phenolic aldehydes from lignin [113]. Usually oxidation depolymerization also involves hydrolysis [64, 110]. The production of vanillin from lignin represent the typical mechanism of the generation of aldehyde group (Fig. 5) [111]. Sun et al. studied the oxidation degradation of wheat straw lignin by alkaline nitrobenzeneand cupric compound (CuO, Cu(OH)2, CuSO4) in detail, and the major products were the phenolic aldehydes of vanillin and syringaldehyde using both of the oxidizing agent [114]. With nitrobenzene and NaOH at 170 ℃, the selectivity of vanillin and syringaldehyde reached 39% and 37%, respectively. It has now been recognized that nitrobenzene is the oxidant that gives the highest yields of the depolymerized phenolic products [18]. However, it is highly toxic and the reduction product (aniline) is difficult to separate. Thus, O2 as a green oxidant is more attractive [65] and has been widely reported in recent years. For example, Chornet et al. conducted the alkaline oxidation of a steam-explosion hardwood lignin in the presence of Cu2+ and Fe2+ using O2 as the oxidant [115], and the yield of combined aldehydes (vanillin + syringaldehyde + hydroxybenzaldehyde) can reach 14.6 wt% with only 1.3 wt% yield of ketone products. Zhu et al. carried out the lignin oxidation reaction in phenol media with a catalyst of γ-Al2O3supported ReOx nanoparticles. The yield of vanillin was 7.3 wt% with selectivity up to more than 95.0% under moderate reaction conditions (120 ℃ and 2 bar O2) [108]. Therein, phenol was used as reaction media to enhance the dissolution of lignin. The pulsed corona discharge (PCD) was applied to lignin oxidation in aqueous solution, which can remove lignin from waste water and transfer it to aldehydes, and the selectivity to aldehydes can be tuned by oxygen concentration [116]. Overall, it is a challenge to obtain complete selectivity to aldehydes by oxidation depolymerization. There's a balance between aldehydes production, aldehydes degradation, and competing reactions, which determines the maximum yield and selectivity [74]. Because of the different reactivity of varies types of lignin and particular structures, the oxidation conditions should be carefully tuned to move the balance for the aldehydes production [74].

|
Download:
|
| Fig. 5. Proposed mechanism for lignin oxidation to generate vanillin by Tarabanko et al. [65, 111]. Adapted with permission [65]. Copyright 2017, Royal Society of Chemistry. | |
{kind=link}
3.3. Carboxylic acids
Carboxylic acids are widely reported as the depolymerization product of lignin, but usually co-exist with other products, such as phenols and aldehydes [104, 117]. They are often considered as "side products", and few works reported them as the main product with high selectivity. Lignin derived carboxylic acid are mainly aromatic acids. The most famous are vanillic acid syringic acid and their derivates, which are the oxidation products of the monomers. However, vanillin and syringaldehyde are more fundamentally important, so the formation of the corresponding acids is not preferred and considered as over oxidation.
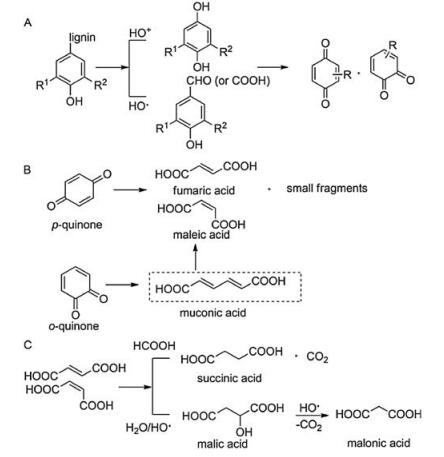
Recently, some researchers attempted to transform lignin into aliphatic acids, such as acetic, muconic, maleic, and succinic acids. The market for this kind of aliphatic acids is much larger than that for aromatic acids and aldehydes [118]. They are important and highly valuable industrial chemicals and intermediates used in biopolymer, pharmaceutical, and food additives industries [119-121]. Currently, they are mainly produced from petroleum-based feedstocks [122]. Producing these important compounds from renewable lignin provides a new alternative and meets the requirement of sustainable development. Zhang et al. reported the selective production of dicarboxylic acids through chalcopyrite(CuFeS2)-catalyzed oxidation of biorefinery lignin including diluted-acid corn stover lignin and steam-exploded spruce lignin using hydrogen peroxide (H2O2) [123]. The total yield of dicarboxylic acids (malonic acid, succinic acid, maleic acidand malic acid) were 14% and 11% for this two lignin samples, respectively, and the selectivity to the dicarboxylic acids was up to 91%. The reaction mechanism is shown in Fig. 6, which involves the depolymerization of lignin, aromatic ring opening and the formation of the final products [123]. Patience and co-workers developed a two-stage gas-solid conversion method that transformed lignin into carboxylic acids [118]. Lignin was thermo-oxidatively steam cracked in the first step to volatile compounds which contact a catalyst bed in the second step to produce carboxylic acids. The products are mostly C4 carboxylic acids such as maleic acid and butyric acid, and the selectivity to different carboxylic acids can be turned by the metal catalyst as well as the support [118] (Fig. 7).

|
Download:
|
| Fig. 6. Mechanisms for lignin depolymerization and aromatic nuclei oxidation (A), aromatic ring cleavage (B), and formation of carboxylic acids (C) [123]. Adapted with permission [123]. Copyright 2017, John Wiley and Sons. | |
{kind=link}

|
Download:
|
| Fig. 7. Highly selective transformation of lignin into acetic acid [66]. Adapted with permission [66]. Copyright 2014, John Wiley and Sons. | |
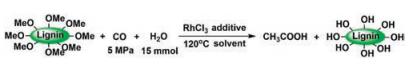{kind=link}
More recently, Han et al. reported a striking result that acetic acid can be selectively obtained from lignin with 100% selectivity (Fig. 7). This is the first time that a single product is harvested from lignin [66]. They proposed a strategy that selective transform a specific group in lignin to value-added chemicals [35]. Applying this strategy, they converted the methoxy groups in lignin into acetic acid as the single product in the liquid phase, while the molecular weight distribution of the residue lignin did not change significantly according to the GPC trace. This method works for both Kraft and organosolv lignin, and the utilization of methoxy group reached up to 87.5%. NMR spectra and HPLC trace verifies that acetic acid is the only product.
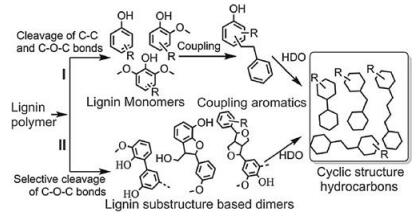
3.4. AlkanesOne of the most significant value of lignin is that it can be converted into bio-oilwhich has the potential to be an alternative to fossil fuels [1]. Despite extensive research, lignin still has a low commercial value and usually is combusted as a low-grade fuel [124]. One of the main distinct differences between bio-oil and fossil fuels is their higher oxygen contents [125], which has many drawbacks such as instability, causticity, high viscosity and low energy density [4, 81, 126]. So, it is desperate to develop methods to decrease oxygen content while increase the H/C ration in the depolymerized liquid product for use of bio-fuel. Because of the similarities to hydrocarbons derived from petroleum, the lignin-derived alkanes could well be refined into drop-in fuels by conventional oil refinery processes. For the catalytic hydrodeoxygenation (HDO) of lignin-derived phenols or bio-oils, please refer to the existing reviews [81, 127-129]. Here we focus on directly depolymerizing lignin into hydrocarbons with high selectivity.
To transform lignin into alkanes, all types of C-O bonds and aromatic rings should be via hydrogenolysis and hydrogenation, as presented in Fig. 8. This requires a very efficient hydrogenolysis and hydrogenation catalyst, and noble metal catalysts are good candidates. Using a two-step process, Kou et al. successfully degraded wood lignin into alkanes over noble metal (Pd/C, Ru/C, Pt/C) catalyzed hydrogenolysis [131]. Lignin was first depolymerized into monomers (200 ℃), which were further hydrogenated into alkanes at higher temperature (250 ℃) in the second step. This approach yielded about 42 wt% C8-C9 alkanes, 10 wt% C14-C18 alkanes, and 11 wt% methanol from lignin, which are close to the empirical maximum values. Ru/Al2O3 combined with acidic zeolite(H+-Y) was also reported to catalyze the HDO of dilute alkali extracted corn stover lignin into jet fuel range hydrocarbons (primarily C12-C18 cyclic structures) [130]. Earth abundant Ni-based catalysts were also applied for the HDO of lignin. For example, Zhao et al. developed an efficient method for one-pot hydrodeoxygenation of enzymatic lignin to C6-C9 cycloalkanes in liquid dodecaneover supported Ni catalyst [132]. The method enables 80 wt% lignin conversions by using amorphous silica-alumina supported Ni particles at 300 ℃ in the presence of 6 MPa H2 with the yield of cycloalkanes approaching 50 wt%, and almost no aromatic compounds generates. The solvent and the acid sites in the support are found to be crucial for the high selectivity. Ni/Al-SBA-15 is also demonstrated to be capable of hydrodeoxygenating organosolv lignin with selectivity to cycloalkanes higher than 99% [133]. Although good achievement has been made on transformation of lignin into liquid alkanes, some researchers are devoted to directly converting raw lignocellulose into liquid fuels. Without chemical pretreatment of raw wood, this strategy has a great potential to be tremendous workload and energy saving [134]. Xia, Chen and co-workers designed a multifunctional Pt/NbOPO4 catalyst that can promote the direct HDO of raw woods into liquid alkanes with mass yields up to 28.1 wt%. Cellulose, hemicelluloses and, more significantly, lignin fractions in the wood sawdust are simultaneously converted into hexane, pentane and alkylcyclohexanes, respectively, which paved an energy-efficient route for converting raw lignocellulose into valuable alkanes [134].

|
Download:
|
| Fig. 8. Routes of depolymerization and hydrodeoxygenation of lignin to cyclic structurehydrocarbons. Adapted with permission [130]. Copyright 2015, Royal Society of Chemistry. | |
{kind=link}
3.5. Arenes
Lignin is the only abundant renewable source for aromatic chemicals. Converting it into liquid alkanes as fuel does not take full use of lignin, because this kind of compounds can be derived from other renewable resource such as cellulose and hemicelluloses. Depolymerization and deoxygenation of lignin while retaining the aromatic ring are very interesting and of great significance, because it provides an inexhaustible supply for arenes which are difficult to be derived from other renewable resource. To achieve this goal, great care should be taken to promote hydrogenolysis of C-O bonds while prevent the hydrogenation of the aromatic rings during the lignin hydrodeoxygenation. But it is extremely challenging because the aryl-O bonds in aryl ethers are strong, especially that in phenol (414 kJ/mol) [135], and the hydrogenation of the aromatic ring is more thermodynamically favoured [45].
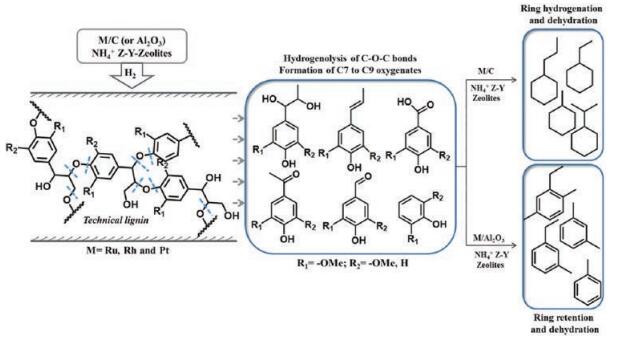
Up to now, there is only a few reports on the highly selective transformation of lignin into aromatic hydrocarbons. Yang et al. developed an aqueous phase catalytic process to selectively depolymerize lignin polymeric framework and remove oxygen via hydrodeoxygenation (HDO) reactions over supported noble metals (Ru, Rh and Pt) in the present of solid acid zeolites. The reaction mechanism is shown in Fig. 9. Under various reaction condition, the conversion of lignin was 35%-60% and the selectivity for aromatic hydrocarbons were 65%-70% [50]. More recently, Yang and Wang et al. reported a one-pot lignin conversion over a Ru/Nb2O5 catalyst in water media [45]. The yield of liquid hydrocarbons was 35.5 wt%, which was a near-quantitative carbon yield based on lignin monomers obtained via nitrobenzene oxidation method. The selectivity for monomer arenes approached 71%. The authors also performed DFT calculations to reveal reaction mechanism, and the results showed that the synergistic effect between Ru and NbOx species contributed to the high selectivity to arenas [45].

|
Download:
|
| Fig. 9. Proposed pathway for formation of aromatic and cyclic aliphatic hydrocarbons from lignins with supported (C and/or Al2O3) noble metals integrated with zeolite NH4+ Z-Y 57277-14-1 catalyst matrix. Adapted with permission [50]. Copyright 2013, Royal Society of Chemistry. | |
{kind=link}
3.6. Other compounds
Other compounds such as ethers [136], esters [137] also appeared as major depolymerization products of lignin in some reports, but the authors did not aim to produce this kind of compounds and there is no data of the selectivity. Substituted cyclohexanols can be obtained from phenolic model compounds with high selectivity [87], but there is no reports for real lignin. Quinones widely exits in oxidation depolymerization products, and there is a few works that yielded it as major product. For example, Bösmann and Wasserscheid et al. found that Mn(NO3)2 can effectively catalyze the oxidation depolymerization of lingin in ionic liquid media, espeically in [EMIM][CF3SO3] (1-ethyl-3-methylimidazolium trifluoromethanesulfonate) [138]. By adjusting the reaction conditions and catalyst loading, 2, 6-dimethoxy-1, 4-benzoquinone (DMBQ) was formed as the main product, which could be isolated from the reaction mixture as a pure substance in 21.0% selectivity and 11.5 wt% overall yield with respect to the amount of lignin [138].
4. Conclusions and outlookIn this minireview, we summarized the selective transformation of lignin into value-added chemicals. Phenols, aldehydes, carboxylic acids, alkanes, arenes et al. can be derived from lignin with relatively high selectivity. However, it is far less than enough. Because we still get mixed products even if the selectivity to a specific class of compounds is 100%! What is worse, the separation of congeners is usually more difficult than different classes of species. There is a lot of work yet to do, because it is the high selective to one single compound that is the ultimate goal for highly efficiently utilization of lignin. But this is extremely challenging because of the complex structure of lignin. Although there are only several main types of linkages in lignin, selective cleavage one specific type of the linkages is a rough ride. Nevertheless, selective cleavage of the linkages does not guarantee the selectivity of the product. Future opportunities for highly selective utilization of lignin:
1) Design more efficient and robust catalyst taking advantages of confinement effects to achieve higher selectivity;
2) Take advantages of the condensation. Although the condensation of the fragments in lignin is usually considered as side reaction which prevents the depolymerization of lignin, it may be helpful for higher selectivity of the depolymerization product. If most of the undesired products could be condensed into larger molecules by carefully controlling the reaction condition, the selectivity to the desired product would be very high and the separation would be easier.
3) Think out of the box and make the selectivity even worse! If lignin can be depolymerized into different classes of compounds and only one product in each class, the separation would be relatively easy and energy saving;
4) Multilevel utilization of lignin. Convert lignin in multistep, and one specific groups is selectively utilized in each step. We only need to achieve high selectivity in each step, and the product can be different from different steps. This will make the separation relatively simple. What is more, different catalysts can be used in each step and they do not need to be multifunctional, which also reduce the challenge of catalyst design.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China(No. 21603235), National Key Research and Development Program of China (No. 2017YFA0403103), and Chinese Academy of Sciences (No. QYZDY-SSW-SLH013).
| [1] |
M. Stöcker, Angew. Chem. Int. Ed. 47 (2008) 9200-9211. DOI:10.1002/anie.200801476 |
| [2] |
A.J. Ragauskas, C.K. Williams, B.H. Davison, et al., Science 311 (2006) 484-489. DOI:10.1126/science.1114736 |
| [3] |
K.K. Lange, E.I. Tellgren, M.R. Hoffmann, T. Helgaker, Science 337 (2012) 327-331. DOI:10.1126/science.1219703 |
| [4] |
D.M. Alonso, S.G. Wettstein, J.A. Dumesic, Chem. Soc. Rev. 41 (2012) 8075-8098. DOI:10.1039/c2cs35188a |
| [5] |
A.J. Ragauskas, G.T. Beckham, M.J. Biddy, et al., Science 344 (2014) 709-719. |
| [6] |
C. Li, X. Zhao, A. Wang, G.W. Huber, T. Zhang, Chem. Rev. 115 (2015) 11559-11624. DOI:10.1021/acs.chemrev.5b00155 |
| [7] |
S.K. Ritter, Chem. Eng. News 85 (2007) 11-17. |
| [8] |
J.D. Gargulak, S.E. Lebo, T.J. McNally, Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., New York, 2001.
|
| [9] |
M. Lewin, I.S. Goldstein, Science 152 (1991) 500-502. |
| [10] |
B. Kamm, P.R. Gruber, M. Kamm, Biorefineries-Industrial Processes and Products, Wiley-VCH Weinheim, 2005.
|
| [11] |
F.G. Calvo-Flores, J.A. Dobado, ChemSusChem 3 (2010) 1227-1235. DOI:10.1002/cssc.v3.11 |
| [12] |
M.P. Pandey, C.S. Kim, Chem. Eng. Technol. 34 (2011) 29-41. DOI:10.1002/ceat.v34.1 |
| [13] |
E. Sjöstrom, Wood Chemistry:Fundamentals and Applications. New York: Academic Press.
|
| [14] |
X.J. Shen, J.L. Wen, P.L. Huang, et al., BioEnergy Res. 10 (2017) 1155-1162. DOI:10.1007/s12155-017-9855-2 |
| [15] |
C.O. Tuck, E. Perez, I.T. Horvath, R.A. Sheldon, M. Poliakoff, Science 337 (2012) 695-699. DOI:10.1126/science.1218930 |
| [16] |
N.M. Belgacem, Monomers, Polymers and Composites from Renewable Resources, Elsevier Ltd., Amsterdam, 2008. https://www.sciencedirect.com/book/9780080453163/monomers-polymers-and-composites-from-renewable-resources
|
| [17] |
M.A.R. Meier, J.O. Metzger, U.S. Schubert, Chem. Soc. Rev. 36 (2007) 1788-1802. DOI:10.1039/b703294c |
| [18] |
M. Fache, B. Boutevin, S. Caillol, ACS Sustain. Chem. Eng. 4 (2016) 35-46. DOI:10.1021/acssuschemeng.5b01344 |
| [19] |
J.H. Lora, W.G. Glasser, J. Polym. Environ. 10 (2002) 39-48. DOI:10.1023/A:1021070006895 |
| [20] |
W. Thielemans, E. Can, S.S. Morye, R.P. Wool, J. Appl. Polym. Sci. 83 (2002) 323-331. |
| [21] |
P.C.A. Bruijnincx, B.M. Weckhuysen, Nat. Chem. 6 (2014) 1035-1036. DOI:10.1038/nchem.2120 |
| [22] |
D. Stewart, Ind. Crops Prod. 27 (2008) 202-207. DOI:10.1016/j.indcrop.2007.07.008 |
| [23] |
W.O.S. Doherty, P. Mousavioun, C.M. Fellows, Ind. Crops Prod. 33 (2011) 259-276. DOI:10.1016/j.indcrop.2010.10.022 |
| [24] |
G. Xu, J.H. Yang, H.H. Mao, Z. Yun, Chem. Technol. Fuels Oils 47 (2011) 283. DOI:10.1007/s10553-011-0297-9 |
| [25] |
N.S. Çetin, N. Özmen, Int. J. Adhes. Adhes. 22 (2002) 481-486. DOI:10.1016/S0143-7496(02)00059-3 |
| [26] |
B.L. Xue, J.L. Wen, R.C. Sun, ACS Sustain. Chem. Eng. 2 (2014) 1474-1480. DOI:10.1021/sc5001226 |
| [27] |
C. Xu, R.A.D. Arancon, J. Labidi, R. Luque, Chem. Soc. Rev. 43 (2014) 7485-7500. DOI:10.1039/C4CS00235K |
| [28] |
J. Zakzeski, P.C.A. Bruijnincx, A.L. Jongerius, B.M. Weckhuysen, Chem. Rev. 110 (2010) 3552-3599. DOI:10.1021/cr900354u |
| [29] |
R. Jastrzebski, S. Constant, C.S. Lancefield, et al., ChemSusChem 9 (2016) 2074-2079. DOI:10.1002/cssc.201600683 |
| [30] |
P.J. Deuss, K. Barta, Coord. Chem. Rev. 306 (2016) 510-532. DOI:10.1016/j.ccr.2015.02.004 |
| [31] |
A. Rahimi, A. Ulbrich, J.J. Coon, S.S. Stahl, Nature 515 (2014) 249-252. DOI:10.1038/nature13867 |
| [32] |
A.K. Deepa, P.L. Dhepe, ACS Catal. 5 (2015) 365-379. DOI:10.1021/cs501371q |
| [33] |
V. Roberts, S. Fendt, A.A. Lemonidou, X. Li, J.A. Lercher, Appl. Catal. B-Environ. 95 (2010) 71-77. DOI:10.1016/j.apcatb.2009.12.010 |
| [34] |
V.M. Roberts, V. Stein, T. Reiner, et al., Chem.-Eur. J. 17 (2011) 5939-5948. DOI:10.1002/chem.v17.21 |
| [35] |
A. Toledano, L. Serrano, J. Labidi, Fuel 116 (2014) 617-624. DOI:10.1016/j.fuel.2013.08.071 |
| [36] |
J. Long, Y. Xu, T. Wang, et al., Appl. Energy 141 (2015) 70-79. DOI:10.1016/j.apenergy.2014.12.025 |
| [37] |
D. Mohan, C.U. Pittman Jr, P.H. Steele, Energy Fuels 20 (2006) 848-889. DOI:10.1021/ef0502397 |
| [38] |
M. Kosa, H. Ben, H. Theliander, A.J. Ragauskas, Green Chem. 13 (2011) 3196-3202. DOI:10.1039/c1gc15818j |
| [39] |
J. Cho, S. Chu, P.J. Dauenhauer, G.W. Huber, Green Chem. 14 (2012) 428-439. DOI:10.1039/C1GC16222E |
| [40] |
P.F. Britt, A.C. Buchanan, K.B. Thomas, S.K. Lee, J. Anal, Appl. Pyrolysis 33 (1995) 1-19. DOI:10.1016/0165-2370(94)00846-S |
| [41] |
F.L.P. Resende, S.A. Fraley, M.J. Berger, P.E. Savage, Energy Fuels 22 (2008) 1328-1334. DOI:10.1021/ef700574k |
| [42] |
F.L.P. Resende, P.E. Savage, AlChE J. 56 (2010) 2412-2420. |
| [43] |
A. Yamaguchi, N. Hiyoshi, O. Sato, M. Shirai, Top. Catal. 55 (2012) 889-896. DOI:10.1007/s11244-012-9857-4 |
| [44] |
G. Chatel, R.D. Rogers, ACS Sustain. Chem. Eng. 2 (2014) 322-339. DOI:10.1021/sc4004086 |
| [45] |
X. Wang, R. Rinaldi, Angew. Chem. Int. Ed. 52 (2013) 11499-11503. DOI:10.1002/anie.201304776 |
| [46] |
X. Wang, R. Rinaldi, Energy Environ. Sci. 5 (2012) 8244-8260. DOI:10.1039/c2ee21855k |
| [47] |
H. Ben, W. Mu, Y. Deng, A.J. Ragauskas, Fuel 103 (2013) 1148-1153. DOI:10.1016/j.fuel.2012.08.039 |
| [48] |
R. Ma, W. Hao, X. Ma, Y. Tian, Y. Li, Angew. Chem. Int. Ed. 53 (2014) 7310-7315. DOI:10.1002/anie.201402752 |
| [49] |
X. Ma, Y. Tian, W. Hao, R. Ma, Y. Li, Appl. Catal. A:Gen. 481 (2014) 64-70. DOI:10.1016/j.apcata.2014.05.002 |
| [50] |
D.D. Laskar, M.P. Tucker, X. Chen, G.L. Helms, B. Yang, Green Chem. 16 (2014) 897-910. DOI:10.1039/c3gc42041h |
| [51] |
L. Shuai, M.T. Amiri, Y.M. Questell-Santiago, et al., Science 354 (2016) 329-333. DOI:10.1126/science.aaf7810 |
| [52] |
K. Barta, G.R. Warner, E.S. Beach, P.T. Anastas, Green Chem. 16 (2014) 191-196. DOI:10.1039/C3GC41184B |
| [53] |
Q. Song, F. Wang, J. Xu, Chem. Commun. 48 (2012) 7019-7021. DOI:10.1039/c2cc31414b |
| [54] |
Q. Song, F. Wang, J. Cai, et al., Energy Environ. Sci. 6 (2013) 994-1007. DOI:10.1039/c2ee23741e |
| [55] |
C. Li, M. Zheng, A. Wang, T. Zhang, Energy Environ. Sci. 5 (2012) 6383-6390. DOI:10.1039/C1EE02684D |
| [56] |
J. Zhang, H. Asakura, J. van Rijn, et al., Green Chem. 16 (2014) 2432-2437. DOI:10.1039/C3GC42589D |
| [57] |
C. Zhao, D.M. Camaioni, J.A. Lercher, J. Catal. 288 (2012) 92-103. DOI:10.1016/j.jcat.2012.01.005 |
| [58] |
S.M. Leckie, G.J. Harkness, M.L. Clarke, Chem. Commun. 50 (2014) 11511-11513. DOI:10.1039/C4CC04939J |
| [59] |
T.L. Marker, L.G. Felix, M.B. Linck, M.J. Roberts, Environ. Prog. Sustain. 31 (2012) 191-199. DOI:10.1002/ep.v31.2 |
| [60] |
T.L. Marker, L.G. Felix, M.B. Linck, et al., Environ. Prog. Sustain. 33 (2014) 762-768. DOI:10.1002/ep.v33.3 |
| [61] |
R. Ma, Y. Xu, X. Zhang, Chemsuschem 8 (2015) 24-51. DOI:10.1002/cssc.201402503 |
| [62] |
F.S. Chakar, A.J. Ragauskas, Ind. Crop. Prod. 20 (2004) 131-141. DOI:10.1016/j.indcrop.2004.04.016 |
| [63] |
A. Vigneault, D.K. Johnson, E. Chornet, Can. J. Chem. Eng. 85 (2007) 906-916. |
| [64] |
J. Luo, P. Melissa, W. Zhao, Z. Wang, Y. Zhu, ChemistrySelect 1 (2016) 4596-4601. DOI:10.1002/slct.201600758 |
| [65] |
P.C. Rodrigues Pinto, E.A. Borges da Silva, A.E. Rodrigues, Lignin as source of fine chemicals: vanillin and syringaldehyde, in: C. Baskar, S. Baskar, R.S. Dhillon (Eds.), Biomass Conversion: The Interface of Biotechnology, Chemistry and Materials Science, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 381-420. https://link.springer.com/chapter/10.1007%2F978-3-642-28418-2_12
|
| [66] |
Q. Mei, H. Liu, X. Shen, et al., Angew. Chem. Int. Ed. 56 (2017) 14868-14872. DOI:10.1002/anie.201706846 |
| [67] |
S.K. Hanson, R.T. Baker, Acc. Chem. Res. 48 (2015) 2037-2048. DOI:10.1021/acs.accounts.5b00104 |
| [68] |
J. Ralph, K. Lundquist, G. Brunow, et al., Phytochem. Rev. 3 (2004) 29-60. DOI:10.1023/B:PHYT.0000047809.65444.a4 |
| [69] |
W. Boerjan, J. Ralph, M. Baucher, Annu. Rev. Plant Biol. 54 (2003) 519-546. DOI:10.1146/annurev.arplant.54.031902.134938 |
| [70] |
R.C. Sun, Cereal Straw As A Resource for Sustainable Biomaterials & Biofuels, Elsevier Oxford, 2010. https://www.sciencedirect.com/book/9780444532343/cereal-straw-as-a-resource-for-sustainable-biomaterials-and-biofuels
|
| [71] |
R. Vanholme, K. Morreel, J. Ralph, W. Boerjan, Curr. Opin. Plant Biol 11 (2008) 278-285. DOI:10.1016/j.pbi.2008.03.005 |
| [72] |
S.D. Mansfield, H. Kim, F. Lu, J. Ralph, Nat. Protocol 7 (2012) 1579-1589. DOI:10.1038/nprot.2012.064 |
| [73] |
J.L. Wen, S.L. Sun, B.L. Xue, R.C. Sun, Materials 6 (2013) 359-391. DOI:10.3390/ma6010359 |
| [74] |
P.C. Rodrigues Pinto, E.A. Borges da Silva, A.E. Rodrigues, Ind. Eng. Chem. Res. 50 (2011) 741-748. DOI:10.1021/ie102132a |
| [75] |
A. Zhang, F. Lu, R. Sun, J. Ralph, Planta 229 (2009) 1099-1108. DOI:10.1007/s00425-009-0894-6 |
| [76] |
X. Shen, P. Huang, J. Wen, R. Sun, Prog. Chem. 29 (2017) 162-180. |
| [77] |
R. Parthasarathi, R.A. Romero, A. Redondo, S. Gnanakaran, J. Phys. Chem. Lett. 2 (2011) 2660-2666. DOI:10.1021/jz201201q |
| [78] |
M.V. Galkin, J.S.M. Samec, ChemSusChem 9 (2016) 1544-1558. DOI:10.1002/cssc.201600237 |
| [79] |
Z. Jiang, H. Zhang, T. He, et al., Green Chem. 18 (2016) 4109-4115. DOI:10.1039/C6GC00798H |
| [80] |
C. Zhao, Y. Kou, A.A. Lemonidou, X. Li, J.A. Lercher, Angew. Chem. Int. Ed. 48 (2009) 3987-3990. DOI:10.1002/anie.v48:22 |
| [81] |
Q. Bu, H. Lei, A.H. Zacher, et al., Bioresour. Technol. 124 (2012) 470-477. DOI:10.1016/j.biortech.2012.08.089 |
| [82] |
F. Liu, Q. Liu, A. Wang, T. Zhang, ACS Sustain. Chem. Eng. 4 (2016) 3850-3856. DOI:10.1021/acssuschemeng.6b00620 |
| [83] |
A.G. Sergeev, J.F. Hartwig, Science 332 (2011) 439-443. DOI:10.1126/science.1200437 |
| [84] |
F. Gao, J.D. Webb, J.F. Hartwig, Angew. Chem. Int. Ed. 55 (2016) 1474-1478. DOI:10.1002/anie.201509133 |
| [85] |
J. He, C. Zhao, J.A. Lercher, J. Am. Chem. Soc. 134 (2012) 20768-20775. DOI:10.1021/ja309915e |
| [86] |
P. Kelley, S. Lin, G. Edouard, M.W. Day, T. Agapie, J. Am. Chem. Soc. 134 (2012) 5480-5483. DOI:10.1021/ja300326t |
| [87] |
V. Molinari, C. Giordano, M. Antonietti, D. Esposito, J. Am. Chem. Soc. 136 (2014) 1758-1761. DOI:10.1021/ja4119412 |
| [88] |
A.G. Sergeev, J.D. Webb, J.F. Hartwig, J. Am. Chem. Soc. 134 (2012) 20226-20229. DOI:10.1021/ja3085912 |
| [89] |
X. Wang, R. Rinaldi, ChemSusChem 5 (2012) 1455-1466. DOI:10.1002/cssc.v5.8 |
| [90] |
E. Adler, Wood Sci. Technol. 11 (1977) 169-218. DOI:10.1007/BF00365615 |
| [91] |
S. Van den Bosch, T. Renders, S. Kennis, et al., Green Chem. 19 (2017) 3313-3326. DOI:10.1039/C7GC01324H |
| [92] |
P. Ferrini, R. Rinaldi, Angew. Chem. Int. Ed. 53 (2014) 8634-8639. DOI:10.1002/anie.201403747 |
| [93] |
X. Huang, O.M. Morales Gonzalez, J. Zhu, et al., Green Chem. 19 (2017) 175-187. DOI:10.1039/C6GC02962K |
| [94] |
X. Huang, J. Zhu, T.I. Korányi, M.D. Boot, E.J.M. Hensen, ChemSusChem 9 (2016) 3262-3267. DOI:10.1002/cssc.v9.23 |
| [95] |
S. Van den Bosch, W. Schutyser, R. Vanholme, et al., Energy Environ. Sci. 8 (2015) 1748-1763. DOI:10.1039/C5EE00204D |
| [96] |
H. Luo, I.M. Klein, Y. Jiang, et al., ACS Sustain. Chem. Eng. 4 (2016) 2316-2322. DOI:10.1021/acssuschemeng.5b01776 |
| [97] |
P.J. Deuss, C.S. Lancefield, A. Narani, et al., Green Chem. 19 (2017) 2774-2782. DOI:10.1039/C7GC00195A |
| [98] |
P.J. Deuss, M. Scott, F. Tran, et al., J. Am. Chem. Soc. 137 (2015) 7456-7467. DOI:10.1021/jacs.5b03693 |
| [99] |
A. Toledano, L. Serrano, A.M. Balu, et al., ChemSusChem 6 (2013) 529-536. DOI:10.1002/cssc.201200755 |
| [100] |
C.S. Lancefield, O.S. Ojo, F. Tran, N.J. Westwood, Angew. Chem. Int. Ed. 54 (2015) 258-262. DOI:10.1002/anie.201409408 |
| [101] |
E. Feghali, G. Carrot, P. Thuéry, C. Genre, T. Cantat, Energy Environ. Sci. 8 (2015) 2734-2743. DOI:10.1039/C5EE01304F |
| [102] |
Z. Jiang, T. He, J. Li, C. Hu, Green Chem. 16 (2014) 4257-4265. DOI:10.1039/C4GC00620H |
| [103] |
L. Hu, Y. Luo, B. Cai, et al., Green Chem. 16 (2014) 3107-3116. DOI:10.1039/C3GC42489H |
| [104] |
M. Wang, J. Lu, X. Zhang, et al., ACS Catal. 6 (2016) 6086-6090. DOI:10.1021/acscatal.6b02049 |
| [105] |
Y. Yang, H. Fan, Q. Meng, et al., Chem. Commun. 53 (2017) 8850-8853. DOI:10.1039/C7CC04209D |
| [106] |
R. Ma, M. Guo, K.T. Lin, et al., Chem.Eur. J. 22 (2016) 10884-10891. DOI:10.1002/chem.201600546 |
| [107] |
A. Rahimi, A. Azarpira, H. Kim, J. Ralph, S.S. Stahl, J. Am. Chem. Soc. 135 (2013) 6415-6418. DOI:10.1021/ja401793n |
| [108] |
E.A.B. de Silva, M. Zabkova, J.D. Araújo, et al., Chem. Engi. Res. Des. 87 (2009) 1276-1292. DOI:10.1016/j.cherd.2009.05.008 |
| [109] |
M.B. Hocking, J. Chem. Educ. 74 (1997) 1055. DOI:10.1021/ed074p1055 |
| [110] |
A.W. Pacek, P. Ding, M. Garrett, G. Sheldrake, A.W. Nienow, Ind. Eng. Chem. Res. 52 (2013) 8361-8372. DOI:10.1021/ie4007744 |
| [111] |
V.E. Tarabanko, D.V. Petukhov, G.E. Selyutin, Kinet. Catal. 45 (2004) 569-577. DOI:10.1023/B:KICA.0000038087.95130.a5 |
| [112] |
A. Azarpira, J. Ralph, F. Lu, BioEnergy Res. 7 (2013) 78-86. |
| [113] |
C. Diaz-Urrutia, W.C. Chen, C.O. Crites, et al., RSC Adv. 5 (2015) 70502-70511. DOI:10.1039/C5RA15694G |
| [114] |
R. Sun, J.M. Lawther, W.B. Banks, Ind. Crop Prod. 4 (1995) 241-254. DOI:10.1016/0926-6690(95)00038-0 |
| [115] |
G. Wu, M. Heitz, E. Chornet, Ind. Eng. Chem. Res. 33 (1994) 718-723. DOI:10.1021/ie00027a034 |
| [116] |
I. Panorel, L. Kaijanen, I. Kornev, et al., Environ. Technol. 35 (2014) 171-176. DOI:10.1080/09593330.2013.821144 |
| [117] |
J. Zeng, C.G. Yoo, F. Wang, et al., ChemSusChem 8 (2015) 861-871. DOI:10.1002/cssc.v8.5 |
| [118] |
S. Lotfi, D.C. Boffito, G.S. Patience, React. Chem. Eng. 1 (2016) 397-408. DOI:10.1039/C6RE00053C |
| [119] |
S.Y. Lee, S.H. Hong, S.H. Lee, S.J. Park, Macromol. Biosci. 4 (2004) 157-164. |
| [120] |
K. Sato, M. Aoki, R. Noyori, Science 281 (1998) 1646-1647. DOI:10.1126/science.281.5383.1646 |
| [121] |
H. Yu, F. Peng, J. Tan, et al., Angew. Chem. Int. Ed. 50 (2011) 3978-3982. DOI:10.1002/anie.201007932 |
| [122] |
A. Castellan, J.C.J. Bart, S. Cavallaro, Catal. Today 9 (1991) 237-254. DOI:10.1016/0920-5861(91)80049-F |
| [123] |
R. Ma, M. Guo, X. Zhang, ChemSusChem 7 (2014) 412-415. DOI:10.1002/cssc.201300964 |
| [124] |
M. Nagy, K. David, G.J.P. Britovsek, A.J. Ragauskas, Holzforschung 63 (2009) 513-520. DOI:10.1515/HF.2009.097 |
| [125] |
M. Schlaf, Dalton T. (2006) 4645-4653. |
| [126] |
C. Zhao, J.A. Lercher, Angew. Chem. Int. Ed. 124 (2012) 6037-6042. DOI:10.1002/ange.201108306 |
| [127] |
D.D. Laskar, B. Yang, H. Wang, J. Lee, Biofuel Bioprod. Bior. 7 (2013) 602-626. DOI:10.1002/bbb.2013.7.issue-5 |
| [128] |
M. Saidi, F. Samimi, D. Karimipourfard, et al., Energy Environ. Sci. 7 (2014) 103-129. DOI:10.1039/C3EE43081B |
| [129] |
P.M. Mortensen, J.D. Grunwaldt, P.A. Jensen, K.G. Knudsen, A.D. Jensen, Appl. Catal. A:Gen. 407 (2011) 1-19. DOI:10.1016/j.apcata.2011.08.046 |
| [130] |
H. Wang, H. Ruan, H. Pei, et al., Green Chem. 17 (2015) 5131-5135. DOI:10.1039/C5GC01534K |
| [131] |
N. Yan, C. Zhao, P.J. Dyson, et al., ChemSusChem 1 (2008) 626-629. DOI:10.1002/cssc.v1:7 |
| [132] |
J. Kong, B. Li, C. Zhao, RSC Adv. 6 (2016) 71940-71951. DOI:10.1039/C6RA16977E |
| [133] |
X. Wang, R. Rinaldi, Catal. Today 269 (2016) 48-55. DOI:10.1016/j.cattod.2015.11.047 |
| [134] |
Q. Xia, Z. Chen, Y. Shao, et al., Nat. Commun. 7 (2016) 11162. DOI:10.1038/ncomms11162 |
| [135] |
Y. Shao, Q. Xia, L. Dong, et al., Nat. Commun. 8 (2017) 16104. DOI:10.1038/ncomms16104 |
| [136] |
X. Huang, T.I. Korányi, M.D. Boot, E.J.M. Hensen, Green Chem. 17 (2015) 4941-4950. DOI:10.1039/C5GC01120E |
| [137] |
J.S. Luterbacher, A. Azarpira, A.H. Motagamwala, et al., Energy Environ. Sci. 8 (2015) 2657-2663. DOI:10.1039/C5EE01322D |
| [138] |
K. Stärk, N. Taccardi, A. Bösmann, P. Wasserscheid, ChemSusChem 3 (2010) 719-723. DOI:10.1002/cssc.v3:6 |