 2018, Vol. 29
2018, Vol. 29 


b State Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, China;
c Key Laboratory of Pesticide & Chemical Biology, Ministry of Education, College of Chemistry, Central China Normal University, Wuhan 430079, China;
d National Research Tomsk Polytechnic University, Tomsk 634050, Russian Federation;
e Hubei University of Technology Engineering and Technology College, Wuhan 430068, China
Cytochrome bc1 complex (complex Ⅲ, EC 1.10.2.2, bc1), one of the five multiprotein complexes (complexes Ⅰ–Ⅴ), are essential components of the respiratory chain [1]. Generally, the bc1 complex is proposed to operate via a Q-cycle mechanism. Based on this mechanism, it can catalyze the electron transfer from ubiquinol to cytochrome c. At the same time, it couples this electron transfer and the translocation of protons across the membrane. Accordingly, a proton gradient and membrane potential are generated to drive ATP synthesis [2-5]. Since the bc1 complex plays a pivotal role in the life process, it has been demonstrated as one of the most attractive targets for numerous pharmaceuticals and agrochemicals. Over the recent years, great attention has been paid to the development of various bc1 complex inhibitors [6-20]. Although numerous inhibitors have been disclosed, discovering active inhibitors with new scaffolds is still highly desirable [21-24].
4-Aryloxyphenyl scaffolds draw our considerable attention since they exhibit promising biological activities, especially inhibitory activities against the bc1 complex [6, 15, 25-27]. For instance, compound A, obtained by structural modifications from the natural product antimycin A, was recognized as a potent inhibitor of the bc1 complex (Fig. 1a) [6, 25]. Moreover, compound B was identified as an excellent bc1 complex inhibitor, and it also displayed superb antimalarial activities (Fig. 1a) [15, 26]. As the continuation of developing new and active bc1 complex inhibitors, we assumed that new structures containing a 4-aryloxyphenyl moiety could be beneficial for discovering new inhibitors of this target. Besides, N-arylphthalimides have attracted our great interest due to their biological and pharmacological activities [28-44]. In addition, the N-arylphthalimide scaffold features chemical diversity which can be achieved from easily accessible building blocks. Therefore, employing a 4-aryloxyphenyl group as the aryl motif of N-arylphthalimides could provide N-(4-aryloxylphenyl)phthalimides, which could be potentially active bc1 complex inhibitors (as shown in Fig. 1b).

|
Download:
|
| Fig. 1. (a) Representative bc1 complex inhibitors A and B containing 4-aryloxyphenyl moieties; (b) N-(4-aryloxylphenyl)phthalimides as potential inhibitors of the bc1 complex. | |
Herein, we planed to synthesize a series of target compounds and evaluate their inhibitory activities against the bc1 complex. To our delight, a few active inhibitors were found after extensive screening. Furthermore, computational simulations were performed to investigate the interaction between a representative inhibitor and the target enzyme. Based on these experimental and theoretical studies, we envisioned that the newly synthesized compounds were presumably inhibitors binding to the Qo site of the bc1 complex.
The conventional method for the synthesis of these compounds was illustrated by reacting 4-aryloxy anilines with phthalic anhydrides [45-53]. Very recently, Jiang and co-workers reported a carbonylation access to phthalimides using self-sufficient directing group and nucleophile [54]. Herein, in order to construct a broad range of N-(4-aryloxyphenyl)phthalimides in a facile and efficient manner, we focused our attention to the easily-accessible standard method. Expectedly, plenty of N-(4-aryloxyphenyl) phthalimides 3 could be afforded by treatment of 4-aryloxy anilines 1 with phthalic anhydrides 2 (as depicted in Scheme 1). It was worth mentioning that the three phenyl rings of 3 were assigned as A, B and C, respectively so as to facilitate our further explanations. Based on the reported procedures [45-47], we identified the optimized reaction conditions as 1 (1.0mmol) and 2 (1.0mmol) in refluxing acetic acid (5.0mL), and were used for further study unless otherwise noted (as illustrated in Table S1 and related description in Supporting information). With the optimized reaction conditions in hand, target products 3a-r were prepared by the reactions of 2a with various 4-aryloxy anilines (Fig. S1 in Supporting information). In addition, the reactions of 1a with various phthalic anhydrides were also explored to yield products 3s-a′ (Table S2 in Supporting information).

|
Download:
|
| Scheme 1. The synthetic route of our designed molecules 3. | |
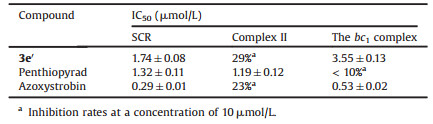
Succinate-cytochrome c reductase (SCR), a mixture of mitochondrial complex Ⅱ (succinate-ubiquinone oxidoreductase) and the bc1 complex, was selected as the target enzyme for initial biochemical evaluation of the above compounds. Several compounds exhibited modest levels of inhibition against SCR at a concentration of 10 μmol/L even though low activities were observed for most compounds, which encouraged us to conduct further structural modifications (Table 1, entries 1–27). We would like to emphasize that the IC50 values of those compounds with inhibitory rates of above 50% at the concentration of 10 μmol/L were further measured (Table 1). It was found that a methyl group at R2 was beneficial for the inhibition potency (entry 27). Thus, R2 was fixed as methyl, and different substituents on R3-R5 were employed to afford compounds 3b′-d′ in excellent yields (entries 28–30). To our delight, 3b′ manifested better inhibitory activities than 3a′ in terms of IC50 values (2.08 μmol/L for entry 28 vs. 6.91 μmol/L for entry 27). It was worth noting that the lowlevels of inhibition for our target molecules were attributed to the poor aqueous solubility in most cases, so a hydrophilic carboxylic group was introduced into the molecular structures to give compounds 3e′-g′ (entries 31–33). Notably, compound 3e′ exhibited good inhibition potency with an IC50 value of 1.74 μmol/L (entry 31). In orderto further confirm whether these compounds were inhibitors of the bc1 complex, the inhibitory activities of representative inhibitor 3e′ against SCR, complex Ⅱ and the bc1 complex were evaluated and shown in Table 2. Expectedly, penthiopyrad and azoxystrobin, the selected two commercial agents, demonstrated excellent activities against complex Ⅱ and the bc1 complex, respectively. Similar with azoxystrobin, 3e′ showed a low inhibition rate against complex Ⅱ, but selective inhibition against the bc1 complex. Therefore, 3e′ has been validated as an inhibitorof the bc1 complex.
|
|
Table 1 Inhibitory activities of the synthesized compounds against porcine SCR. |
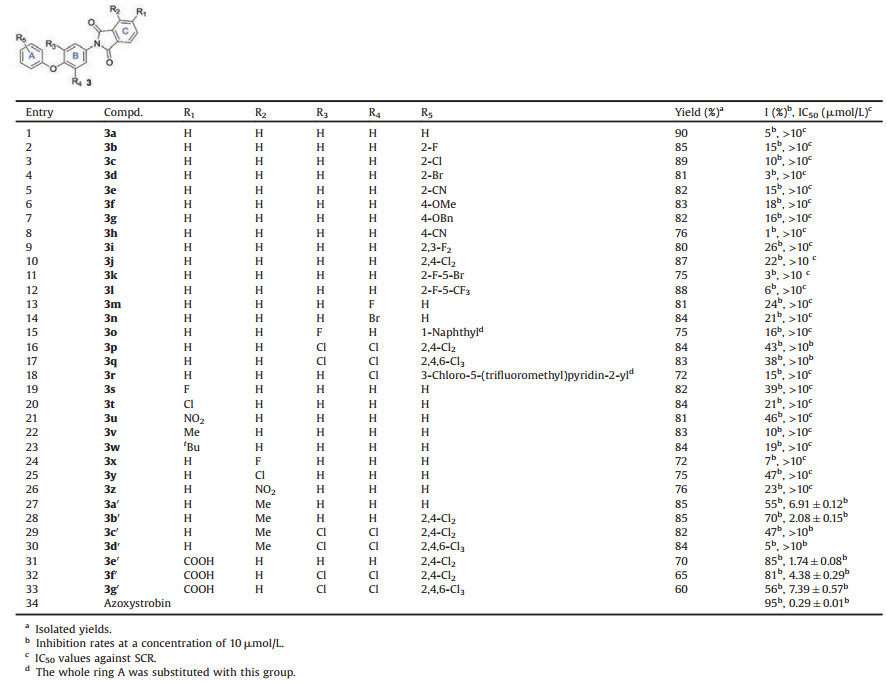
|
|
Table 2 The inhibition effects of the selected inhibitors against SCR, complex Ⅱ and the bc1 complex. |
{kind=link}
{kind=link}
It has been well-documented that the bc1 complex has two quinol/quinone binding sites (Qo and Qi sites) on opposite sides of the membrane [55-57]. Therefore, with an aim to further ascertain the binding siteof these inhibitors, representative inhibitor 3e′ was docked into both Qo and Qi sites to obtain the respective 3e′-Qo and 3e′-Qi complexes. When 3e′ was docked into the Qo site, the calculated binding free energy was -8.80kcal/mol for the best binding conformation by using the semi-empirical score function in AutoDock 4.0. Nevertheless, the binding free energy was calculated as -6.57kcal/mol if it bound to the Qi site (Table S3 in Supporting information). Moreover, 10ns molecular dynamics (MD) simulations were obtained for both complexes, and the 100 snapshots achievedfrom the last 1ns of each complex were used to calculate the binding free energy. As shown inTable S3, the binding free energy of 3e′-Qo and 3e′-Qi complexes were calculated as -9.48 and -1.03kcal/mol using the molecular mechanics/PoissonBoltzmann surface area (MM/PBSA) method in Amber16 program, respectively. Therefore, these results suggested that 3e′ might bind to the Qo site of the bc1 complex.
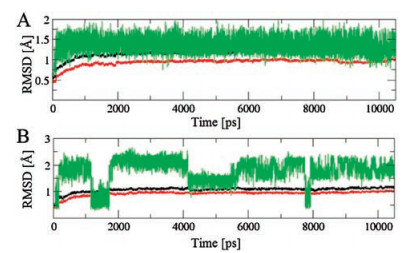
Except for the above calculations, plot of root-mean-square deviation (RMSD) of the protein backbone and inhibitor atoms throughout the whole process of MD simulation were examined from the MD trajectories to evaluate the stability of the 3e′-Qo and 3e′-Qi complexes (Fig. 2). Besides, molecular docking experiments for both binding sites were also done and depicted in Fig. 3. The results implied that the 3e′-Qo complex was very stable within the applied simulation time (Fig. 2A). For the docking of 3e′ into the Qo site, it formed an H-bond with key residue E272, and hydrophobic interactions with F275, F129, F151, I147, L195 and P271 (Fig. 3A) However, obvious fluctuation was clearly spotted in the MD trajectory of the 3e′-Qi complex (Fig. 2B), which was probably due to its instablity. Moreover, 3e′ formed weak π-π hydrophobic interactions with F18 and F220 of the Qi site in Fig. 3B. In order to evaluate the reliability of our docking results, azoxystrobin (the commercial Qo-site inhibitor used as a control agent for bioassays) and famoxadone (a commercial Qo-site inhibitor which bears a 4-aryloxyphenyl group) were docked into the Qo site (as shown in Figs. 3C and E, respectively). In addition, an overlay of 3e′ and azoxystrobin or famoxadone in the Qo site was also demonstrated in Fig. 3D or F. These results indicated that 3e′, azoxystrobin and famoxadone formed H-bonds with the key residue E272. However, these two commercial agents could form more hydrophobic interactions with active site residues than 3e′. On the basis of all the above results, it was proposed that 3e′ should be a Qo-site inhibitor.

|
Download:
|
| Fig. 2. Time dependence of the RMSD of the whole protein atoms (color in black), protein backbone atoms (color in red) and inhibitors (color in green) (A) 3e′-Qo and (B) 3e′-Qi complexes. | |
{kind=link}

|
Download:
|
| Fig. 3. (A) The binding mode of 3e′ in the Qo site. (B) The binding mode of 3e′ in the Qi site. (C) The binding mode of azoxystrobin in the Qo site (PDB ID: 3L71). (D) The overlay of 3e′ and azoxystrobin in the Qo site. (E) The binding mode of famoxadone in the Qo site. (F) The overlay of 3e′ and famoxadone in the Qo site. The red dashed lines in (A), (B), (C) and (E) represent the H-bond distances between an inhibitor and a residue. In addition, the H-bond distances between 3e′ or azoxystrobin and the residue were depicted in red and blue dashed lines of (D), respectively; the H-bond distances between 3e′ or famoxadone and the residue were depicted in red and blue dashed lines of (F), respectively. | |
{kind=link}
In summary, a new series of N-(4-aryloxyphenyl)phthalimides with various substituents were synthesized via a simple and efficient method. With these newly prepared compounds in hand, their inhibitory activities against SCR were evaluated. To our delight, a few compounds exhibited good inhibition potency against SCR. Especially, compound 3e′ was found to be the most potent inhibitor, and it was further confirmed to inhibit exclusively the activity of the cytochrome bc1 complex. Furthermore, it has been well-known that the bc1 complex contains two binding sites, so computational simulations were utilized to clarify the binding site of our compounds. The results indicated that representative inhibitor 3e′ probably bound to the Qo site of the bc1 complex. Hopefully, this work could be of great value for the development of other bc1 complex inhibitors.
AcknowledgmentsThis research was supported by the National Natural Science Foundation of China (Nos. 21502062, 21272091 and 21472063), Hubei Provincial Department of Education (No. Q20102606), Xiangyang Science and Technology Bureau (No. 2010GG1B33), and Structural Biomedicine and Pharmacochemistry of Hubei University of Arts and Science. Francis Verpoort acknowledges the support from the Russian Foundation for Basic Research (No. 18-29-04047) and the Tomsk Polytechnic University Competitiveness Enhancement Program grant (No. VIU-195/2018).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2018.10.008.
| [1] |
H. Cheng, Y.Q. Shen, X.Y. Pan, et al., New J. Chem. 39 (2015) 7281-7292. DOI:10.1039/C5NJ00215J |
| [2] |
H. Kim, D. Xia, C.A. Yu, et al., Proc. Natl. Acad. Sci. U. S. A. 95 (1998) 8026-8033. DOI:10.1073/pnas.95.14.8026 |
| [3] |
E.A. Berry, M. Guergovakuras, L.S. Huang, A.R. Crofts, Annu. Rev. Biochem. 69 (2000) 1005-1075. DOI:10.1146/annurev.biochem.69.1.1005 |
| [4] |
A.R. Crofts, Annu. Rev. Physiol. 66 (2004) 689-733. DOI:10.1146/annurev.physiol.66.032102.150251 |
| [5] |
D. Xia, L. Esser, L. Yu, C.A. Yu, Photosynth. Res. 92 (2007) 17-34. DOI:10.1007/s11120-007-9155-3 |
| [6] |
S. Bolgunas, D.A. Clark, W.S. Hanna, P.A. Mauvais, S.O. Pember, J. Med. Chem. 49 (2006) 4762-4766. DOI:10.1021/jm060408s |
| [7] |
A.G. Kruglov, M.A. Andersson, R. Mikkola, et al., Chem. Res. Toxicol. 22 (2009) 565-573. DOI:10.1021/tx800317z |
| [8] |
E.A. Berry, L.S. Huang, D.W. Lee, et al., Biochim. Biophys. Acta Bioenerget. 1797 (2010) 360-370. DOI:10.1016/j.bbabio.2009.12.003 |
| [9] |
P.L. Zhao, L. Wang, X.L. Zhu, et al., J. Am. Chem. Soc. 132 (2010) 185-194. DOI:10.1021/ja905756c |
| [10] |
F. Wang, H. Li, L. Wang, et al., Bioorg. Med. Chem. 19 (2011) 4608-4615. DOI:10.1016/j.bmc.2011.06.008 |
| [11] |
C. Vallieres, N. Fisher, T. Antoine, et al., Antimicrob. Agents Chemother. 56 (2012) 3739-3747. DOI:10.1128/AAC.00486-12 |
| [12] |
W.C. Yang, H. Li, F. Wang, X.L. Zhu, G.F. Yang, ChemBioChem 13 (2012) 1542-1551. DOI:10.1002/cbic.v13.11 |
| [13] |
X. Zhu, F. Wang, H. Li, et al., Chin. J. Chem. 30 (2012) 1999-2008. DOI:10.1002/cjoc.201200607 |
| [14] |
G.F. Hao, F. Wang, H. Li, et al., J. Am. Chem. Soc. 134 (2012) 11168-11176. DOI:10.1021/ja3001908 |
| [15] |
M.J. Capper, P.M. O'Neill, N. Fisher, et al., Proc. Natl. Acad. Sci. U. S. A. 112 (2015) 755-760. DOI:10.1073/pnas.1416611112 |
| [16] |
X.L. Zhu, M.M. Zhang, J.J. Liu, J.M. Ge, G.F. Yang, J. Agric. Food Chem. 63 (2015) 3377-3386. DOI:10.1021/acs.jafc.5b00228 |
| [17] |
M. Fehr, A. Wolf, G. Stammler, Pest Manag. Sci. 72 (2016) 591-602. DOI:10.1002/ps.2016.72.issue-3 |
| [18] |
C. Chen, Q.Y. Wu, L.Y. Shan, et al., RSC Adv. 6 (2016) 97580-97586. DOI:10.1039/C6RA19424A |
| [19] |
P.H. Alday, I. Bruzual, A. Nilsen, et al., Antimicrob. Agents Chemother. 61 (2017) e01866-16. |
| [20] |
Z. Song, B.I. Iorga, P. Mounkoro, N. Fisher, B. Meunier, FEBS Lett. 592 (2018) 1346-1356. DOI:10.1002/1873-3468.13035 |
| [21] |
H. Cheng, Q.Y. Wu, F. Han, G.F. Yang, Chin. Chem. Lett. 25 (2014) 705-709. DOI:10.1016/j.cclet.2014.03.013 |
| [22] |
F. Zhang, D. Wu, G.L. Wang, et al., Chin. Chem. Lett. 28 (2017) 1044-1048. DOI:10.1016/j.cclet.2016.12.014 |
| [23] |
J. Chen, F. Sun, P. Chen, et al., Chin. Chem. Lett. 29 (2018) 1151-1154. DOI:10.1016/j.cclet.2018.04.005 |
| [24] |
Y. Zhang, X. Peng, H. Ren, et al., Chin. Chem. Lett. 29 (2018) 719-723. DOI:10.1016/j.cclet.2017.09.053 |
| [25] |
N. Tokutake, H. Miyoshi, H. Nakazato, H. Iwamura, Biochim. Biophys. Acta 1142 (1993) 262-268. DOI:10.1016/0005-2728(93)90154-8 |
| [26] |
C.L. Yeates, J.F. Batchelor, E.C. Capon, et al., J. Med. Chem. 51 (2008) 2845-2852. DOI:10.1021/jm0705760 |
| [27] |
H. Cheng, W. Song, R. Nie, et al., Bioorg. Med. Chem. Lett. 28 (2018) 1330-1335. DOI:10.1016/j.bmcl.2018.03.014 |
| [28] |
J.M. Chapman Jr, P.J. Voorstad, G.H. Cocolas, I.H. Hall, J. Med. Chem. 26 (1983) 237-243. DOI:10.1021/jm00356a022 |
| [29] |
M.D. Coutinho Neto, W.C. Pinheiro Filho, B.B. Neto, M.N. Ramos, J. Braz. Chem. Soc. 4 (1993) 139-142. DOI:10.5935/0103-5053.19930030 |
| [30] |
V. Bailleux, L. Vallee, J.P. Nuyts, J. Vamecq, Biomed. Pharmacother. 47 (1994) 463-464. |
| [31] |
Y. Shibata, K. Sasaki, Y. Hashimoto, S. Iwasaki, Biochem. Biophys. Res. Commun. 205 (1994) 1992-1997. DOI:10.1006/bbrc.1994.2904 |
| [32] |
Y. Shibata, K. Sasaki, K. Nishimura, Y. Hashimoto, S. Iwasaki, Biol. Pharm. Bull. 17 (1994) 1532-1534. DOI:10.1248/bpb.17.1532 |
| [33] |
A.B. Cruz, R.C.B. Cruz, V. Cechinel Filho, et al., Rev. Latinoam. Quim. 25 (1996) 10-13. |
| [34] |
Y. Shibata, K. Sasaki, Y. Hashimoto, S. Iwasaki, Chem. Pharm. Bull. 44 (1996) 156-162. DOI:10.1248/cpb.44.156 |
| [35] |
H. Sano, T. Noguchi, A. Tanatani, H. Miyachi, Y. Hashimoto, Chem. Pharm. Bull. 52 (2004) 1021-1022. DOI:10.1248/cpb.52.1021 |
| [36] |
W. Pluempanupat, S. Adisakwattana, S. Yibchok-Anun, W. Chavasiri, Arch. Pharmacal Res. 30 (2007) 1501-1506. DOI:10.1007/BF02977317 |
| [37] |
S. Mangani, L. Cancian, R. Leone, et al., J. Med. Chem. 54 (2011) 5454-5467. DOI:10.1021/jm2005018 |
| [38] |
C.I. Manley-King, J.J. Bergh, J.P. Petzer, Bioorg. Med. Chem. 19 (2011) 4829-4840. DOI:10.1016/j.bmc.2011.06.070 |
| [39] |
V. Kathuria, D.P. Pathak, Pharma Innov. 1 (2012) 55-59. |
| [40] |
A. Aliabadi, B. Gholamine, T. Karimi, Med. Chem. Res. 23 (2014) 2736-2743. DOI:10.1007/s00044-013-0870-3 |
| [41] |
S.P.O. Assis, T.G. Araujo, V.L.M. Sena, et al., Med. Chem. Res. 23 (2014) 708-716. DOI:10.1007/s00044-013-0673-6 |
| [42] |
O. Takahashi, R. Kirikoshi, N. Manabe, Int. J. Mol. Sci. 16 (2015) 12174-12184. DOI:10.3390/ijms160612174 |
| [43] |
R. Caraballo, M. Larsson, S.K. Nilsson, et al., Eur. J. Med. Chem. 103 (2015) 191-209. DOI:10.1016/j.ejmech.2015.08.058 |
| [44] |
J.J. Casal, M. Bollini, M.E. Lombardo, A.M. Bruno, Eur. J. Pharm. Sci. 83 (2016) 114-119. DOI:10.1016/j.ejps.2015.12.017 |
| [45] |
S.K. Dolui, B.K. Mandal, S. Maiti, Angew. Makromol. Chem. 189 (1991) 51-61. DOI:10.1002/apmc.1991.051890105 |
| [46] |
M.D. Shau, W.K. Chin, J. Polym. Sci. Part A:Polym. Chem. 31 (1993) 1653-1658. DOI:10.1002/pola.1993.080310702 |
| [47] |
R.J. Perry, S.R. Turner, R.W. Blevins, Macromolecules 27 (1994) 4058-4062. DOI:10.1021/ma00093a005 |
| [48] |
Y.T. Hong, S.C. Kim, K.Y. Choi, Polym. Bull. 42 (1999) 661-668. DOI:10.1007/s002890050516 |
| [49] |
T.V. Holland, T.E. Glass, J.E. McGrath, Polymer 41 (2000) 4965-4990. DOI:10.1016/S0032-3861(99)00578-9 |
| [50] |
J.K. Im, J.C. Jung, Polymer 41 (2000) 8709-8716. DOI:10.1016/S0032-3861(00)00266-4 |
| [51] |
Z.M. Liang, H. Yin, H.H. Xu, Polymer 44 (2003) 1391-1399. DOI:10.1016/S0032-3861(02)00911-4 |
| [52] |
C. Gao, X. Wu, G. Lv, M. Ding, L. Gao, Macromolecules 37 (2004) 2754-2761. DOI:10.1021/ma0350720 |
| [53] |
M. Evecen, G. Duru, H. Tanak, A.A. Agar, J. Mol. Struct. 1118 (2016) 1-9. DOI:10.1016/j.molstruc.2016.03.079 |
| [54] |
F. Ji, J. Li, X. Li, et al., J. Org. Chem. 83 (2018) 104-112. DOI:10.1021/acs.joc.7b02433 |
| [55] |
S. Iwata, J.W. Lee, K. Okada, et al., Science 281 (1998) 64-71. DOI:10.1126/science.281.5373.64 |
| [56] |
D. Xia, H. Kim, C.A. Yu, et al., Biochem. Cell Biol. 76 (1998) 673-679. DOI:10.1139/o98-090 |
| [57] |
X. Gong, L. Yu, D. Xia, C.A. Yu, J. Biol. Chem. 280 (2005) 9251-9257. DOI:10.1074/jbc.M409994200 |