 2018, Vol. 29
2018, Vol. 29 

Numerous devastating cell-degenerative diseases such as Alzheimer's disease (AD) have been identified to be linked to deposition of amyloid formed by the self-assembly of proteins. AD is a common senile dementia, which is pathologically sourced from the accumulation of intracellular neurofibrillary tangles of tau protein [1] and extracellular senile plagues of amyloid β (Aβ) [2]. The amyloid plagues are considered to be formed by four steps, (1) Aβ containing 39–43 residues is produced by sequential cleavage of the amyloid precursor protein (APP) by both β- and γ- secretases [3, 4], Aβ42 are considered as most toxic form; (2) Aβ monomers with enriched β-sheet structure are prone to form oligomers and protofibrils, linking to the neuron dysfunction [5]; (3) the oligomers and protofibrils further aggregate to form nanofibers (NFs); (4) NFs accumulate into amyloid plaques. It is accepted that soluble Aβ oligomers and protofibrils are most toxic species. Therefore, inhibiting aggregation [6] of Aβ displays certain beneficial effects on AD by several inhibitors, such as small molecules [7], antibodies [8], and nanoparticles (NPs) [9].
Self-assembled NPs, the formation of which is driven by weak interactions, such as hydrophobic/hydrophilic, hydrogen bonding, and electrostatic interactions similar to protein folding, membrane forming, etc., are structurally bioinspired nanomaterials [10-15]. More importantly, self-assembled NPs with sophisticated surface may mimic functionality of protein and interact with other biomolecules/proteins by controlling the weak interactions [16, 17]. For example, self-assembled NPs mimic the functions of natural molecular chaperones [18] (Hsp70, apolipoprotein E (apoE)) to control undesired protein unfolding by binding to exposed hydrophobic residues [19]. From the viewpoint of biomimic, we would like to design a chaperone protein-like NPs that can recognize and co-assemble with Aβ to inhibit the formation of toxic species of self-assembled Aβ.
Peptide sequence of Lys-Leu-Val-Phe-Phe (KLVFF) as a segment of Aβ, which is considered as the main driver of the fibrillation of Aβ, can target Aβ and inhibit the aggregation through co-assembly by strong H-bonding and hydrophobic interactions [20, 21]. We recently focused on self-assembly and the structural transformation of bis(pyrene)-KLVFF based peptides, especially in specific physiological/pathological conditions [23-25]. The BP-KLVFF could self-assemble into NPs, followed by transformation into nanofibers (NFs), which could be induced by ligand-receptor interactions or hydrophobic/hydrophilic interactions. Moreover, the studies on BP-KLVFF-poly ethylene glycol (PEG) indicate that longer hydrophilic chains may induce faster transformation [26, 27]. It is well known that KLVFF is a key segment of Aβ for the formation of toxic Aβ assemblies. Therefore, the BP-KLVFF based NPs were expected to be as artificial chaperones to recognize and co-assemble with Aβ due to the KLVFF sequence.
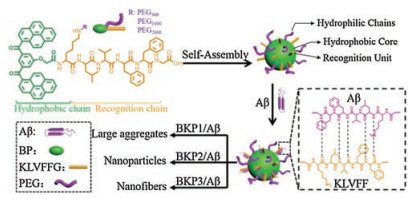
Herein, we prepared a series of BP-KLVFFG-PEG (BKP) molecules and systematically explored how NPs co-assembled with Aβ42, inhibiting the formation of Aβ42 toxic self-assembled species and the toxicity on neuron. BKP1, BKP2, and BKP3 were named corresponding to molecular weight of 368, 1000, and 2000 of PEG, respectively, self-assembled into NPs, which could recognize and capture Aβ42 through H-bonding and hydrophobic interactions of KLVFF unit. The different length of PEG chain as outer shell endowed BKP1, BKP2 and BKP3 with different morphology of co-assemblies including large aggregates, NPs, NFs, probably resulting in different capture efficiencies. BKP2 and Aβ42 co-assembled into particulate structures, exhibiting highest capture efficiency (68%), dramatic decreasing in the cytotoxicity of Aβ42 to neuron, which may be ascribed to the decrease of Aβ42 deposition on cell surfaces (Scheme 1).

|
Download:
|
| Scheme 1. Molecular structures of the peptide with different PEG chain (Mw = 368, 1000, 2000), forming self-assembled NPs, which capture and co-assemble with Aβ42 into large aggregates, nanoparticles, and nanofibers, respectively | |
{kind=link}
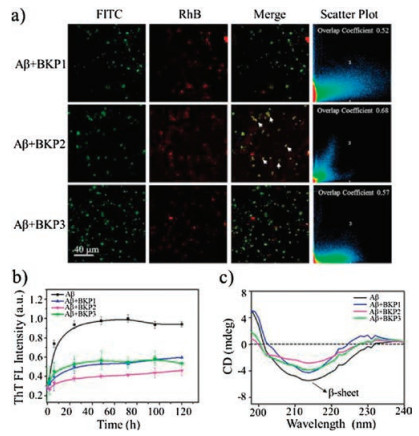
We started with synthesis of molecules BKPn and named as BKP1, BKP2, BKP3 with PEG (Mw = 368, 1000, 2000), bearing BP as hydrophobic unit linked to KLVFFG peptide sequence at the Nterminus of main chain and N-terminus of side chain of Lys residue as attachment sites for PEG units. The NPs with 2.0 × 10-5 mol/L in H2O with 1% DMSO in volume were prepared and the diameter were about from 30 nm to 100 nm [22]. The NPs were composed by three units, hydrophobic core, hydrophilic shell and KLVFF as target units recognizing and capturing Aβ42. The critical micelle concentration (CMC) values of BKPn and Aβ42 obtained by quantitative analysis of fluorescence emission of BP with concentrations varies were lower than that of BKPn, indicating coassembly of Aβ42 by BKPn NPs (Figs. S1 and S2 in Supporting information). In order to further confirm the recognition and coassembly of Aβ42 by BKPn NPs, we carried out the co-localization of BKPn NPs and Aβ42 by confocal laser scanning microscopy (CLSM) measurement. FITC was used to label Aβ42 with green fluorescence. BKPn NPs were labeled with Rhodamine B (RhB) to form BKP1-RhB, BKP2-RhB and BKP3-RhB with red emission. Fortunately, the fluorescence from BKPn NPs with maximum emission at 520 nm can be quenched due to labelled RhB when excited at 350 nm due to fluorescence resonance energy transfer. Therefore, from the fluorescence imaging, the BKPn and Aβ42 could be located with different fluorescence, respectively. CLSM observation of cocultured BKPn-RhB and Aβ42-FITC for 4 h showed that red fluorescence from RhB was merged with green fluorescence from FITC, indicating the co-localization of BKPn and Aβ42. These results suggested BKPn NPs recognize and co-assemble with Aβ42 through KLVFF for capture of Aβ42. Moreover, the co-localization coefficient (0.68, defined as capture efficiency) between BKP2 and Aβ42 was higher than those of BKP1/Aβ42 (0.52) and BKP3/Aβ42 (0.57) (Fig. 1a), indicating that the capture efficiency of Aβ42 by BKPn NPs was affected by PEG chain length, which would be discussed below.

|
Download:
|
| Fig. 1. The capture of Aβ42 by BKPn NPs. (a) Confocal laser scanning microscope (CLSM) imaging of co-culture of Aβ42-FITC (1.0 × 10-5 mol/L), BKP1-RhB (1.0 × 10-5 mol/L), BKP2-RhB (1.0 × 10-5 mol/L) and BKP3-RhB (1.0 × 10-5 mol/L). The white arrow indicated the co-localization of BKPn and Aβ42. (b) Time-dependent ThT fluorescence changes for Aβ42 (2.0 × 10-5 mol/L) incubated with different BKPn NPs (1.0 × 10-5 mol/L). ThT fluorescence of Aβ42 aggregates without BKPn was normalized. (c) The CD spectra of Aβ42 (2.0 × 10-5 mol/L) incubated with BKPn NPs at 120 h | |
{kind=link}
The co-assembly of BKPn and Aβ42 was expected to inhibit Aβ42 self-aggregation. To further confirm the co-assembly of BKPn and Aβ42, Thioflavin T (ThT) fluorescence assay which is a widely used dye-binding method to monitor Aβ42 aggregation process through increasing fluorescence intensity, was applied to detect the inhibitory effect of Aβ42 self-aggregation by BKPn NPs. As shown in Fig. 1b, the aggregation of Aβ42 was obviously inhibited by BKPn NPs. The ThT fluorescence profile showed an almost negligible lag phase, a fast growth phase within 24 h, and a steady equilibrium phase after 24 h for 2.0 × 10-5 mol/L Aβ42 alone at 37 ℃. The fluorescence intensity of neat Aβ42 was high due to the large amount of Aβ42 fibrillation formation. However, ThT fluorescence of Aβ42 in the presence of BKPn-RhB NPs increased slowly and finally reached low fluorescence intensity, indicating the small amount of Aβ42 fibrillation, maybe due to the recognition and coassembly by BKPn NPs. BKP2 showed more obvious inhibition of Aβ42 aggregation with the higher inhibition ratio (52.3%, the calculation method was shown in supporting information) than that of BKP1 (42.9%) and BKP3 (42.3%) treated Aβ42.
Circular dichroism (CD) spectra were also utilized to further confirm the inhibition of Aβ42 aggregation by BKPn NPs (Fig. 1c). The Aβ42 showed typical β-sheet structure with 215 nm negative bands after culturing 24 h. BKP1, BKP2 and BKP3 cultured Aβ42 showed decreased intensity at 215 nm compared with Aβ42 alone in 24 h, which revealed the inhibition effect of Aβ42 fibrillation from the capture of BKPn NPs. Similarly, BKP2 group showed the lower intensity at 215 nm than that of BKP1 and BKP3 group. These results were consistent with the fluorescence co-localization of BKPn and Aβ42, indicating the BKP2 binding to and co-assembling with Aβ42 with highest efficiency.
In order to obtain the insight of the co-assembly of Aβ42 and BKPn NPs, transmission electron microscopy (TEM) were used for detecting superstructure of co-assemblies. The fresh solutions of BKPn NPs (2.0 × 10-5 mol/L) were incubated with Aβ42 (2.0 × 10-5 mol/L) in PBS solution at 37 ℃ for 24 h, respectively, and Aβ42 alone (2.0 × 10-5 mol/L) as a control. As shown in Fig. 2, the Aβ42 alone formed NPs with diameter of average 8 ± 2 nm at 1 h and further aggregated into NFs with diameter about 5 ±1 nm at 24 h. However, BKP2 + Aβ42 showed main species of particulate structures (62 ± 6 nm) at 1 h, and turned into larger ones (161 ±19 nm) after 24 h, which may be ascribed to the co-assembly of BKP2 NPs and Aβ42. The KLVFF in BKP2 NPs recognized and coassembled with Aβ42, which could be stabilized by the hydrophilic chain PEG1000 to form large co-assembled NPs. In contrast, BKP1 + Aβ42 showed particulate structures with diameter of about 74 ±7 nm at 1 h and large aggregates at 24 h. The morphology transformation from NPs to large aggregates was probably due to the co-assembly with Aβ42, which increased the hydrophobicity of surfaces of BKP1 NPs. The co-assemblies of BKP1 and Aβ42 cannot be further stabilized by PEG368, leading to the formation of large aggregation with hydrophobic interactions. BKP3 NPs were prone to NFs because of long hydrophilic PEG2000 chains. As a result, the Aβ42 incubated BKP3 formed NPs with diameter of 69 ± 6 nm at 1 h and transformed into short NFs of BKP3 and Aβ42 co-assemblies with diameter of 14 ± 3 nm at 24 h, which was different from Aβ42 alone. For both BKP1 and BKP3 incubated Aβ42 with super structural transformation, the amount of active KLVFF (in outer layer of superstructures) units would decrease upon transformation, which may explain the lower capture efficiency of BKP1 and BKP3 than that of BKP2. To verify whether BKPn NPs were able to modulate Aβ42-induced cell death, the cytotoxicity of Aβ42 on the neuron SH-SY5Y was evaluated using the CCK-8 assay. Firstly, Fig. S2 showed the BKPn NPs were not cytotoxic for SH-SY5Y cells when the concentration was up to 4.0 × 10-5 mol/L. Then, the cell viability of SH-SY5Y cells incubated with BKPn + Aβ42 was carried out under the biocompatible concentration of BKP.

|
Download:
|
| Fig. 2. Structure of co-assembly of BKPn and Aβ42. TEM images of BKPn (2.0 × 10-5 mol/L) nanoaggregates cultured with Aβ42 (2.0 × 10-5 mol/L) at 1 h and 24 h | |
{kind=link}
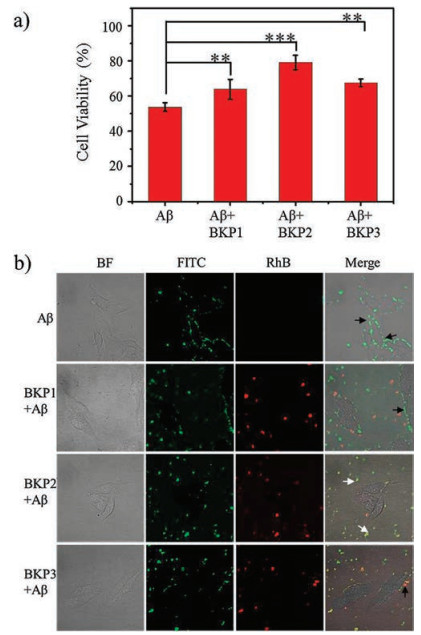
As shown in Fig. 3a, pure Aβ42 (2.0 × 10-5 mol/L) cultured human neuroblastoma SH-SY5Y cells showed a significant cytotoxicity with cell viability of 53.7% relative to the untreated control group. In presence of BKPn NPs (2.0 × 10-5 mol/L), cell viability of Aβ42 (2.0 × 10-5 mol/L) treated SH-SY5Y cells was obviously increased. Moreover, for BKP2 and Aβ42 co-cultured group, cell viability was increased up to 79.1%, which was higher than that of BKP1 (63.8%) and BKP3 (67.6%) (Fig. 3a). The experimental results indicated that capture of Aβ42 by BKPn NPs led to the decrease of toxicity of Aβ42. The higher capture efficiency was, the lower cytotoxicity of Aβ42 was.

|
Download:
|
| Fig. 3. The anti-toxicity of Aβ42 by BKPn. (a) Cytotoxicity of human neuroblastoma SH-SY5Y induced by BKP1, BKP2, BKP3 and Aβ42 using CCK-8 assay. In the CCK-8 assay, the cell viability treated with PBS buffer alone was set as 100%. The cells were cultured with BKPn and Aβ42 at 37 ℃ for 24 h. The concentration of Aβ42 was 2.0 × 10-5 mol/L and the concentration of BKP1, BKP2, and BKP3 were 2.0 × 10-5 mol/L. (b) CLSM imaging of SH-SY5Y cells that were incubated with BKPn and Aβ42 for 4 h. The black arrow indicated the position of Aβ42 on the surface of cell and the white arrow denoted Aβ42 captured by BKPn, blocking Aβ42 from cell surface | |
{kind=link}
In order to give the insight how the capture of Aβ42 by BKPn NPs affected the cytotoxicity, we carefully observed the neuron cultured by Aβ42 (2.0 × 10-5 mol/L) and BKPn (2.0 × 10-5 mol/L) NPs by the CLSM. The SH-SY5Y cells were incubated in Aβ42-FITC, the mixture of Aβ42-FITC and BKP1-RhB, Aβ42-FITC and BKP2-RhB, and Aβ42-FITC and BKP3-RhB for 4 h, respectively. The results were shown in Fig. 3b, the Aβ42-FITC treated SH-SY5Y cells exhibited green fluorescence signals on the cell surface, indicating that Aβ42 might get stuck in the membrane during internalization into cells due to the hydrophobicity, which probably led to neurotoxicity to SH-SY5Y cells. Interestingly, the SH-SY5Y cells treated with Aβ42- FITC/BKPn-RhB NPs showed obviously different results. BKPn NPs significantly decrease the Aβ42 accumulation on cell surfaces due to the co-assembly, which entered into the cells carrying Aβ42. BKP1 and BKP3 with relative lower capture efficiency treated groups still exhibited weak green Aβ42-FITC fluorescence on cell surfaces. However, BKP2 with higher capture efficiency showed almost no Aβ42-FITC on cell surfaces, probably leading to lowest toxicity of Aβ42 to SH-SY5Y cells.
In summary, the KLVFF-functionalized NPs could recognize and co-assemble with Aβ42 through KLVFF recognition group. The resulting co-assemblies showed different morphologies, such as large aggregates, NPs and NFs corresponding to the different length chain of PEG, which showed different capture efficiency and cytotoxicity. The co-assemblies of BKPn and Aβ42 could change the interactions between Aβ42 and cells by decreasing the deposition of Aβ42 on cell membrane, which may be the reason of reduction of Aβ42 to cells. These findings provide the biomimic strategy of material preparation for inhibition of Aβ42 self-aggregation, potentially for AD therapy.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 51573031, 21374026 and 51573032), the National Science Fund for Distinguished Young Scholars (No. 51725302), the Science Fund for Creative Research Groups of the National Natural Science Foundation of China (No. 11621505) and CAS Key Research Program for Frontier Sciences (No. QYZDJ-SSWSLH022). Key Project of Chinese Academy of Sciences in Cooperation with Foreign Enterprises (No. GJHZ1541) and CAS Interdisciplinary Innovation Team.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, atdoi:https://doi.org/10.1016/j.cclet.2018.10.003.
| [1] |
R.A. Rissman, W.W. Poon, M. Blurton-Jones, et al., J. Clin. Invest. 114 (2004) 121-130. DOI:10.1172/JCI200420640 |
| [2] |
B.A. Chromy, R.J. Nowak, M.P. Lambert, et al., Biochem. 42 (2003) 12749-12760. DOI:10.1021/bi030029q |
| [3] |
T. Iwatsubo, Curr. Opin. Neurobiol. 14 (2004) 379-383. DOI:10.1016/j.conb.2004.05.010 |
| [4] |
A.M. Saraiva, I. Cardoso, M.J. Saraiva, et al., Macromol. Biosci. 10 (2010) 1152-1163. DOI:10.1002/mabi.200900448 |
| [5] |
G. Bitan, S.S. Vollers, D.B. Teplow, J. Biol. Chem. 278 (2003) 34882-34889. DOI:10.1074/jbc.M300825200 |
| [6] |
C. Cabaleiro-Lago, F. Quinlan-Pluck, I. Lynch, et al., J. Am. Chem. Soc. 130 (2008) 15437-15443. DOI:10.1021/ja8041806 |
| [7] |
G.C. Look, J. Jerecic, D.B. Cherbavaz, et al., Curr. Alzheimer Res. 4 (2007) 562-567. DOI:10.2174/156720507783018271 |
| [8] |
Y. Du, R. Dodel, H. Hampel, et al., Neurology 57 (2001) 801-805. DOI:10.1212/WNL.57.5.801 |
| [9] |
C.T. Middleton, P. Marek, P. Cao, et al., Nat. Chem. 4 (2012) 355-360. DOI:10.1038/nchem.1293 |
| [10] |
L. Wang, L.L. Li, Y.S. Fan, H. Wang, Adv. Mater. 25 (2013) 3888-3898. DOI:10.1002/adma.v25.28 |
| [11] |
A.P. Xu, P.P. Yang, C. Yang, et al., Nanoscale 8 (2016) 14078-14083. DOI:10.1039/C6NR03580A |
| [12] |
P. Yang, L. Wang, H. Wang, Chin. J. Chem. 33 (2015) 59-70. DOI:10.1002/cjoc.v33.1 |
| [13] |
K. Zhang, P.P. Yang, J.P. Zhang, L. Wang, H. Wang, Chin. Chem. Lett. 28 (2017) 1808-1816. DOI:10.1016/j.cclet.2017.07.001 |
| [14] |
L. Wang, L.L. Li, H.L. Ma, H. Wang, . Chin. Chem. Lett. 24 (2013) 351-358. DOI:10.1016/j.cclet.2013.03.018 |
| [15] |
P.P. He, X.D. Li, L. Wang, H. Wang, J. Mol. Sci. 34 (2018) 89-102. |
| [16] |
A.E. Nel, L. Maedler, D. Velegol, et al., Nat. Mater. 8 (2009) 543-557. DOI:10.1038/nmat2442 |
| [17] |
M. Lundqvist, J. Stigler, G. Elia, et al., P. Natl. Acad. Sci. U. S. A. 105 (2008) 14265-14270. DOI:10.1073/pnas.0805135105 |
| [18] |
F. Huang, J. Wang, A. Qu, et al., Angew. Chem. Int. Ed. 53 (2014) 8985-8990. DOI:10.1002/anie.201400735 |
| [19] |
B. Le Droumaguet, J. Nicolas, D. Brambilla, et al., ACS Nano 6 (2012) 5866-5879. DOI:10.1021/nn3004372 |
| [20] |
S.M. Chafekar, H. Malda, M. Merkx, et al., ChemBioChem 8 (2007) 1857-1864. |
| [21] |
Q. Luo, Y.X. Lin, P.P. Yang, et al., Nat. Commun. 9 (2018) 1802. DOI:10.1038/s41467-018-04255-z |
| [22] |
P.P. Yang, X.X. Zhao, A.P. Xu, L. Wang, H. Wang, J. Mater. Chem. B 4 (2016) 2662-2668. DOI:10.1039/C6TB00097E |
| [23] |
P.P. Yang, Q. Luo, G.B. Qi, et al., Adv. Mater. 29 (2017) 1605869. DOI:10.1002/adma.201605869 |
| [24] |
P.P. Yang, X.X. Zhao, A.P. Xu, L. Wang, H. Wang, J. Mater. Chem. B 4 (2016) 2662-2668. DOI:10.1039/C6TB00097E |
| [25] |
X.X. Hu, P.P. He, G.B. Qi, et al., ACS Nano 11 (2017) 4086-4096. DOI:10.1021/acsnano.7b00781 |
| [26] |
W. Li, P.P. Yang, L. Wang, H. Wang, J. Mater. Chem. C 3 (2015) 3783-3789. DOI:10.1039/C4TC02987A |
| [27] |
L. Wang, W. Li, J. Lu, et al., J. Phys. Chem. C 117 (2013) 26811-26820. DOI:10.1021/jp409557g |