 2018, Vol. 29
2018, Vol. 29 

b State Key Laboratory of New Ceramics and Fine Processing, Advanced Materials of Ministry of Education of China, Tsinghua University, Beijing 100084, China;
c National Center for NanoScience and Technology, Beijing 100190, China
Since highly ordered mesoporous silica materials were first reported by the Mobil company in 1992 [1-3], mesoporous silica nanoparticles (MSNs) have attracted considerable interest among researchers in the fields of catalysis, biomolecule separation, and biomedicine. This interest has been garnered because of their continuously adjustable apertures of 2–50 nm in width, ordered pore structures, narrow pore size distributions, specific high surface areas, and excellent biocompatibility [4-8]. In particular, since the 2001 report by Vallet-Regi et al. in which mobil composition of matter (MCM)-41-type MSNs were first used as a drug delivery system, research on biomedical applications of MSNs as drug delivery systems has increased dramatically [8]. Numerous approaches towards the preparation of different structures comprised of MSNs having uniform cylindrical pores with tunable diameters in the 2–30 nm range have been reported, such as ordered mesoporous silica obtained through self-assembly [9, 10], hollow structural MSNs achieved via the soft-template method [11-13], and the hollow/rattle-type by self-template method [14, 15] or selective etching strategy [16-18]. However, most of these works have focused on the design of MSNs with traditional morphologies and structures, such as spherical coreshell structural and hexagonal MSNs on the nanometer scale. In contrast, the synthesis of mesoporous materials with broad structural length scales from the nano- to micro-scale and exhibiting extraordinary structures has rarely been reported [19, 20]. Therefore, the development of facile methods for the synthesis of micro-scale MSNs with unique pore structures is still highly welcome, considering their broad potential for drug delivery applications.
Recently, several research groups have reported realization of a special intricate polymer silica composite architecture via deactivation enhanced atom transfer radical polymerization (DE-ATRP) [21]. Further exploration of the DE-ATRP approach to generate a polymer replica having complex three-dimensional (3D) structures with controlled features at multiple scales may yield a 3D structure potentially facilitating a new, cost-effective technique for micro-scale particle assembly in the field of biomaterials. In 2015, a diatom biosilica having a hierarchical arrangement of 3D pore patterns with diameters in the nano- to micro-scale range (20 nm– 2 μm) was successfully used as a carrier of both antibodies and hydrophobic drugs for active targeting cancer chemo-therapy [22]. However, the production and efficiency of biomimetic synthesis are the main limitations hindering the application of this biosilica. Meanwhile, the preparation process is more complex and timeconsuming than the artificial synthesis [23]. Therefore, it is meaningful to design and develop a generic, simple, highefficiency method for the synthesis of micro-scale MSNs.
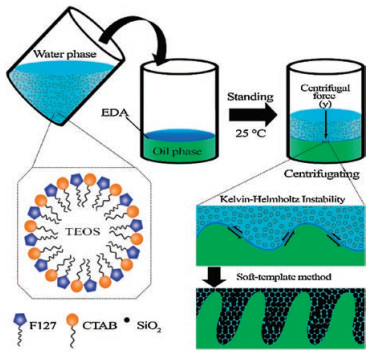
With this motivation, a facile approach to preparing a new hierarchical sieve-like structure of MSN aggregates (hsMNA) using a simple centrifugal method at room temperature was realized for the first time in this study. The specific components of this synthesis process are cetyltrimethyl ammonium bromide (CTAB) and poloxamer (F127) as the template, tetraethyl orthosilicate (TEOS) as the silica source, ethylene diamine (EDA) as the catalyst, and dichloromethane (DCM)-octane as the co-solvent system. Most importantly, the entire procedure can be completed within a short period of time, facilitating high-volume production of microscale hsMSNA with high yield, high surface area, uniform pore distribution, and excellent biocompatibility. The proposed centrifugal method constitutes a new cost-effective technique for microscale hierarchical particle assembly in the field of biomaterials.
Before the preparation, CTAB (≥99%), F127, TEOS (reagent grade, 98%) and concentrated hydrochloric acid (ACS reagent, 37%) were purchased from Sigma-Aldrich LLC. (St. Louis, Missouri, USA). EDA, ethanol, DCM, and n-octane were bought from Sinopharm Group Co., Ltd. (Beijing, China). Deionized water (DI) was obtained from Sinopharm Chemical Reagent Beijing Co., Ltd. (Beijing, China). Roswell Park Memorial Institute (RPMI) 1640 medium (RPMI 1640), fetal bovine serum (FBS), penicillin-streptomycin (PS), and trypsin were bought from Gibco Life Technologies (Beijing, China). Cell proliferation and cytotoxicity assay kits (CCK-8 assay) were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, Jiangsu, China). All other chemicals and reagents were of analytical grade from commercial sources.
To prepare the hsMSNA, we first synthesized colloidal silicon dioxide as the precursor. Specifically, 4 g of F127 and 1 g of CTAB were dissolved in a mixture of DI (5 mL) and anhydrous ethanol (10 mL) at room temperature, and then stirred for 1 h until the solution was clear. Next, 15 mL of TEOS was added to the solution, followed by 2 mL of concentrated hydrochloric acid. After another 2 h of stirring, the precursor was obtained by leaving the final solution to stand at ambient temperature for a designated time. The hsMSNA were prepared in accordance with the following steps. First, 24 mL of DCM and 6 mL of n-octane were placed in a 50-mL centrifugation tube to ensure a reaction liquid density of 1.2 g/mL; then, 0.5 mL of EDA was added. After 4 mL of colloidal silicon dioxide precursor was added to the reactor, sieve-like mesoporous silica was obtained through centrifugation at 15, 000 rpm and 20 ℃ for 5 min. To remove the CTAB and F127, the products were heated to 550 ℃ at a rate of 10 ℃/min and kept at 550 ℃ for 4 h in an air atmosphere. After cooling, the final hsMSNA were obtained.
The morphologies, structures, and pore sizes of the mesoporous silica and hsMSNA were measured using scanning electron microscopy (SEM; S-4800, Hitachi, Japan) and transmission electron microscopy (TEM; HT700, HITACHI, Japan). For the TEM observation, calcined samples were dispersed in ethanol (99.9 vol%) using the ultrasonic method, and the suspension was subsequently dropped onto a carbon microgrid for further testing. The N2 adsorption-desorption isotherms were obtained on a Quantachrome Autosorb-1MP instrument (ASAP 2020, USA) at 77 K. All samples were outgassed at 180 ℃ for 4 h prior to the measurements. Surface area calculations were performed using the Brunauer-Emmett-Teller (BET) and Barrett-Joyner-Halanda (BJH) equation fitted to the first 10 points of each previously obtained isotherm. In addition, the pore size distributions were calculated using Schmidt's modification of the Kelvin equation to account for mono- and multi-layer adsorption. X-ray diffraction (XRD) powder patterns were measured on a Rigaku Multiplex Model D/Max-3c X-ray diffractometer operated at 2 kW and using Cu Kα radiation, in the range of 3°–60° (2θ); the data was collected on a high-resolution diffractometer located at the Siberian Center of Synchrotron Radiation.
L929 mouse fibroblast cells were acquired from the American Type Culture Collection (ATCC) and cultured in RPMI-1640 supplemented with 10% FBS and 1% penicillin and streptomycin for in vitro cytotoxicity. All cells were cultured in humidified incubators under 37 ℃ with 5% CO2. The cytotoxicity of the synthesized hsMSNA was investigated using the L929 cells. Specifically, the L929 cells were detached, counted, and seeded in a 48-well plate at a concentration of 1 ×104 cells/mL after reaching confluence. After 24-h incubation, the original medium was extracted (by sucking) and 0.4 mL of silica dispersion solution was added to each well of the plate. Here, different hsMSNA (sterilized with 60Co gamma ray irradiation) concentrations of 200, 100, 50, 20 and 0 μg/mL were used. After incubation for a designated time, the cell viability was tested via CCK-8 assay at 24, 48, and 72 h. For the CCK-8 assay, after all the original medium had been extracted from the 48-well plate, 200 mL of prepared CCK-8 solution was added to each hole of this plate. After 2 h incubation, 100 μL of solution was extracted and then added to a new 96-well plate. The number of cells was measured with a microplate reader at a 450 nm wavelength.
DOX was loaded into the hsMSNA as follows. Specifically, 0.1 mg DOX was added into 10 mL hsMSNA in PBS (5 mg/mL) at pH 7.4 and stirred at 37 ℃ for 24 h. Then the free DOX was removed via centrifugation and as-synthesized DOX-loaded hsMSNA were stored at 4 ℃. A dialysis method was conducted to assess the release profiles of DOX from DOX-loaded hsMSNA. Briefly, the weighted DOX-loaded hsMSNA were added into a pretreated dialysis bag (MWCO 4000 Da) and then the dialysis bag was placed into 50 mL of release medium (PBS, pH 7.4) at 37 ℃ and stirred at a shaking bath. At predetermined time intervals, 0.5 mL of the incubated medium was withdrawn to measure the amount of DOX using a microplate reader at 475 nm and then equal volume of fresh PBS was supplemented. The release profiles were conducted in triplicate.
The sieve-like structure formation mechanism was analyzed and a schematic illustration of the hsMSNA preparation process is shown in Scheme 1. After dissolution in a mixture of ethanol and water, CTAB and F127 formed the micelle through assembly due to the hydrophobic interaction. The as-synthesized colloidal silica, which is taken as the key source of mesoporous silica to form the hsMSNA, was formed after adding the TEOS and hydrochloric acid (HCl). Before centrifugation, the colloidal silica was placed at 25 ℃ for a designated time to facilitate generation of a homogenous oilwater interface between DCM/n-octane (oil phase) and DI water (water phase), namely, the EDA layer. The final sieve-like structure was formed based on the Kelvin-Helmholtz instability [24] and soft-template method [25] during the centrifugal process.

|
Download:
|
| Scheme 1. Suggested mechanisms illustrating synthesis process of sieve-like structure of MSNs (hsMSNA) by centrifugation | |
Specifically, during the centrifugation process, the density (ρ) and velocity potential (U) of water phase and oil phase are different, and the static interface will move with a velocity 
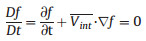
|
where f(x,y,t)=0 presents the perturbed interface. Note that y- η(x, t)=0 at every point (x, y) on the interface at any time. Therefore, the kinematic boundary condition applies, which states that the interface moves up and down with a velocity equal to the vertical component of the fluid velocity from the centrifugal force. The dynamic boundary condition (ϕ) is derived from the unsteady Bernoulli equation and the pressure is continuous across the interface. Thus, the final steady-flow relation in the interface is described as follows:
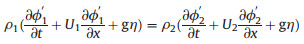
|
here, due to the effect of η(x, t), which stems from the centrifugal force, the interface between the water and oil phases is distorted and a portion of the oil phase flows into the water phase, forming the transition. Different EDA fractions and centrifugal forces directly affect the transition radius, yielding the final sieve-like, mesh-like, and vesicular-like hsMSNA structure. Further, the formation of the transition in the interface layer is the key process to producing cylindrical pores under the centrifugal force.
Meanwhile, the as-formed micelles induced by the CTAB and F127 in the water phase act as the soft template for the MSN growth. Briefly, the EDA in the interface layer diffuses into the micelles, thereby accelerating the TEOS hydrolysis. The TEOS hydrolysis presented in the water phase could produce a large number of negatively charged silicate molecules. Furthermore, the CTAB cationic surfactant, together with F127, easily connects with the silicate molecules via electrostatic attraction. Finally, under the alkaline environment formed by the EDA, the condensation of the self-assembled silicate in the available space among the radical CTAB induces crystallization of the colloidal silica. The hsMSNA sieve-like structure consisting of 40–100 nm MSNs is obtained; then, the soft template is removed through the calcination of the materials at 550 ℃ in the air atmosphere.
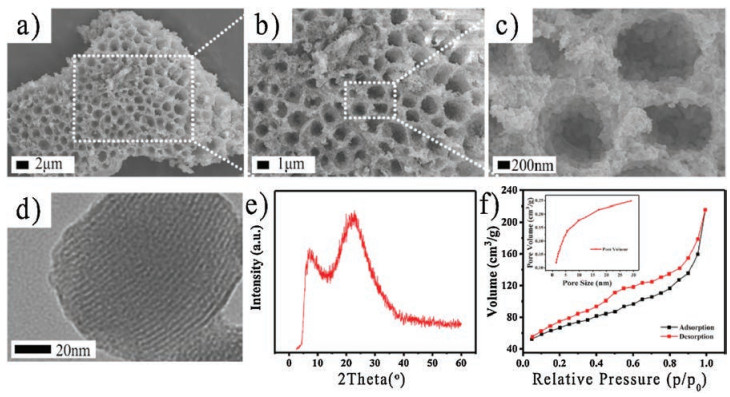
The morphologies of the as-synthesized hsMSNA were measured using both SEM and HRTEM; the results are shown in Fig. 1. The hsMSNA, which were relatively long at ~40 μm, were selfassembled and comprised of uniform small MSNs (Figs. 1a–c) with diameters of 40–100 nm (Fig. 1d). The sieve-like structures were formed when MSNs assembled along the distorted transition induced by the Kelvin-Helmholtz instability. In the vertical direction, the accumulation hallmark for the obtained hsMSNA is the hierarchical arrangement of 3D patterns of cylindrical pores with diameters in the nano- to microscale range (1–2 μm) (Fig. 1c). Fig. 1e showed the XRD patterns of the hsMSNA and a broad diffraction peak appeared at 2θ ≈ 23° corresponded to the amorphous peak of SiO2, which indicates that hsMSNA was in the amorphous state [26]. Fig. 1f displayed the N2 adsorption-desorption isotherms and the corresponding pore-volume distribution curve for hsMSNA. The hsMSNA exihibited a type-Ⅳ isotherm with a H3-type hysteresis loop (p/p0 > 0.2), indicating the presence of cylindrical pores and hierarchical structure. The hierarchical pore structure and the characteristics of the pores themselves indicate that the hsMSNA will exhibit high surface areas [22, 27].

|
Download:
|
| Fig. 1. (a, b, c) Scanning electronic microscopy (SEM) images of hsMSNA under different magnifications; (d) HRTEM image of MSNs comprising hsMSNA; XRD pattern (e) and N2 adsorption (black) and desorption (red) patterns (f) of hsMSNA. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article) | |
In order to successfully synthesize hsMSNA with uniform and controllable particle sizes and pores, various conditions were established by changing the chemical volumes to obtain different hsMSNA. The results show that the EDA acted as a good sizecontrol agent by influencing the acidity of the reaction situation. Therefore, we performed a systematic investigation of the effects of varying EDA fractions on the sieve-like structures of the hsMSNA. The sizes, morphologies, and structures of hsMSNA synthesized using different EDA fractions were investigated using SEM and TEM (Fig. 2). When the EDA volume was increased from 0.5 mL to 4 mL, different sieve-like structures and pore sizes were obtained. In particular, differences in the hierarchical structures were observed in specific directions. In the vertical direction, sieve- (Figs. 2a1–a3), mesh- (Figs. 2b1–b3), and vesicular-like (Figs. 2c1–c3) 3D structures corresponding to 0.5, 2, and 4 mL EDA, were obtained, respectively. We assume that, with increasing EDA content, the interface viscosity is increased. Moreover, the higher alkaline environment induced by the EDA accelerates the TEOS hydrolysis rate, during which the deposition speed is considerably larger than the growth rate, finally yielding a vesicular-like structure. N2 adsorption-desorption isotherms of different hsMSNA exhibited the same typical Ⅳ isotherm but different desorption path and volume value due to the network-percolation effects and a variation of pore diameter along single channels [28, 29]. It was clearly that mesh-like structure resulted a highest adsorption amount because of the more complicated pore distribution.

|
Download:
|
| Fig. 2. SEM, TEM images and N2 adsorption-desorption isotherms of different hsMSNA. EDA content: (a1–a4)0.5ml; (b1-b4)2ml; and(c1-c4)4ml | |
During preparation, the colloidal growth standing time and the centrifugation speed are other main factors influencing the formation of the sieve-like structures of the hsMSNA. For 0.5 mL EDA and an 18 h standing time, the centrifugation time was decreased from 10 to 5 min; hence, it was found that the hsMSNA exhibit a higher surface area (477.10 m2/g) and larger pore volume (0.47 m3/g) (Table 1) for a lower centrifugation rate, which produces a lower force. Further, the average hsMNA pore diameter increases from 3.98 nm to 4.14 nm as the centrifugation time is extended, because there is more time for the colloidal silica assembly process. The high pore volumes of the hsMSNA indicate that they could be endothermic and, thus, constitute a promising material for use in drug delivery systems, catalysis, and sewage purification [30-32].
|
|
Table 1 Surface areas and pore volumes of silica particles with different centrifugation times |

{kind=link}
{kind=link}
{kind=link}
Furthermore, the influence of the colloidal growth standing time under different centrifugation rates was considered (Fig. 3). When the colloidal growth standing time was 8 h, no hsMSNA could be formed, even under a high centrifugation rate of 15, 000 rpm. In that case, the sieve-like structures did not form as the oil-water interface formation was incomplete (Figs. 3a and c). Therefore, no transition process occurred. However, after the standing time was increased to 18 h, a sieve-like structure was observed. Further, the sieve-like structure was affected by the centrifugation speed (Figs. 3b and d). That is, with increasing centrifugation speed, the morphology and microstructure of the sieve-like structure was better, resulting in the increased pore diameters and better uniformity of pores. This enhancement occurred because a larger curvature of distortion is obtained under a larger centrifugation-induced force.

|
Download:
|
| Fig. 3. SEM images of MSN aggregates obtained under different centrifugation rates and standing times | |
{kind=link}
In order to evaluate the structures and surface properties of hsMSNA with and without pores, Brunauer-Emmett-Teller (BET) and nitrogen adsorption-desorption measurements were performed. The corresponding nitrogen adsorption-desorption isotherms, pore-size distribution curves, and Fourier transform infrared (FT-IR) spectra of the samples are shown in Figs. 4a and b, respectively. The hsMSNA both with and without pores exhibited a type-Ⅳ isotherm with a H3-type hysteresis loop in the relative pressure range of 0.5–1.0 (Fig. 4a), implying the presence of variously sized, slit-shaped mesoporous structures. However, the area of the type-Ⅳ isotherm with the H3-type hysteresis loop for the sample with pores is larger than that for the sample without pores. Further, the BJH desorption pore volume curve for the hsMSNA without pores consists of a relatively sharp peak (3 nm), whereas the curve for the hsMSN with pores consists of a relatively sharp peak (3 nm), a feeble shoulder peak (10 nm), and a broad peak (20 nm, Fig. 4b). Moreover, the hsMSNA with pores has a high diameter of 176 nm, confirming the multimodal pore size distributions. Finally, the pore volumes are similar, as the hysteresis loop is consistent with the large pore size of the hsMSNA. To further characterize the differences between the hsMSNA with or without pores, FT-IR analysis was conducted. Fig. 4c shows these results. The peak at 1061 cm-1 in the curve corresponds to the asymmetric stretching vibration of the siloxane (-Si-O-Si-) group, and the bands at 769 and 456 cm-1 can be assigned to the symmetric stretching and deformation modes of this group. In addition, the stretching and bending vibrations of the -OH functional group are apparent at wavenumbers of 3387 and 1570 cm-1, respectively. All these characteristic peaks of hsMSNA with or without pores can be assigned to SiO2, and both exhibit the same surface properties. The nanostructures of the hsMSNA with and without pores were further determined through XRD analysis (Fig. 4d). Further confirmation was achieved by examining the small-angle XRD patterns, in which there are no clear characteristic crystalline diffraction peaks. However, for both sample types, the peak at 2θ = 23° corresponds to a characteristic dispersion peak of amorphous silica, and the low intensity of the peak indicates that both kinds of as-synthesized MSN aggregates substantially consist of disordered amorphous silica. However, the peak intensity is higher for the as-synthesized hsMSNA with pores, suggesting that this sample type has a finer structure.

|
Download:
|
| Fig. 4. (a) N2 adsorption (black and blue) and desorption (red and green) patterns of silica nanoparticle aggregates without (black and red) and with (blue and green) large pores; Inset: Pore size distribution of silica nanoparticle aggregates with (red) or without (black) pores. (b) Particle diameter distribution curves of silica nanoparticle aggregates with (red) or without (black) pores. Inset: Diameter distribution in 0–35 nm range. (c) FT-IR spectra of MSN aggregates with (red) or without (black) pores. (d) XRD patterns of as-synthesized MSN aggregates with (red) or without (black) pores. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article) | |
{kind=link}
In recent years, mesoporous silica is widely used in drug delivery systems in the field of nanomedicine due to its high surface areas and significant biocompatibility [33-36]. Therefore, the biocompatibility or the cytotoxicity of silica has a critical influence on its further applications. Before the in vitro cytotoxicity measurement, the stability of hsMSNA in RPMI-1640 medium was evaluated and the results (Figs. 5a and b) revealed that the hierarchical sieve-like structure of hsMSNA kept same after 72 h incubation. The compatibilities of hsMSNA with different concentrations (20, 50, 100, 200 μg/mL) were evaluated by co-incubating the particles with L929 cells for a certain period of time. The cell viability was evaluated through CCK-8 assay and the results (Fig. 5c) indicate that the difference in cell viabilities after 24- and 48 h incubation was negligibly small. Further, there was little cytotoxicity after incubation for concentrations up to 200 μg/mL, which indicates that the hsMSNA could act as potential carriers for biomedicine application [37].

|
Download:
|
| Fig. 5. The SEM images of hsMSNA before (a) and after (b) incubation with RPMI-1640 medium for 72 h. (c) Effects of hsMSNA with different concentrations on L929 cell viability, as measured through CCK-8 assay | |
{kind=link}
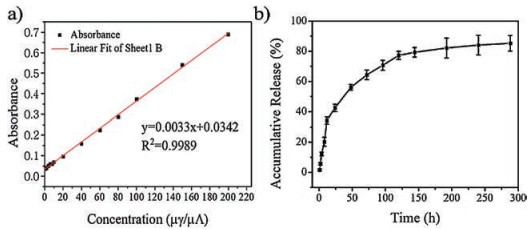
To determine hsMSNA as a drug delivery vehicles, in vitro drug release was conducted under a physiological environment, in PBS at 37 ℃ [38]. The loading efficiency of DOX in hsMSNA, which defined as the ratio of the weight of DOX loaded into the hsMSNA to the total weight of the feeding drugs, was 79.5%. The high encapsulation efficiency may be attribute to the hierarchical pore structure of hsMSNA which will block the transporting of DOX molecule in mesopores. Fig. 6a was the calibration curve of DOX made by a microplate reader at 475 nm. The release kinetics of DOX from hsMSNA demonstrated burst release behaviors at first 24 h and a sustained release pattern at pH 7.4 (Fig. 6b). Finally, approximately 84% of total DOX was release. Considering the excellent biocompatibility of hsMSNA, the above results indicated hsMSNA could be a valuable drug delivery system for nanomedince [39-42].

|
Download:
|
| Fig. 6. (a) The calibration curve of DOX measured by microplate reader. (b) The accumulative release profiles of DOX from DOX-loaded hsMSNA | |
{kind=link}
In summary, we successfully synthesized hsMSNA with different structures through a simple centrifugal method at room temperature, which is based on Kelvin-Helmholtz instability and the softtemplate method. Characterization results revealed that the synthesized hsMSNA consist of 40–100 nm MSNs, piled around 1– 2 μm cylindrical pores in sieve-like tissues. Moreover, different preparation conditions were examined to determine the optimal synthetic process (0.5 mL EDA, standing for 18 h, and centrifugingfor 5 min at 15, 000 rpm). BET and nitrogen adsorption-desorption measurements indicated that the hsMSNA have a high specific surface area of 500m2/g with a pore volume of 0.47 cm3/g.Moreover, the EDA fraction and the standing time for colloidal silica formation are the key factors to influence the hsMSN morphology and structure. Finally, because of the excellent biocompatibility, high drug encapsulation efficiency and sustained release property, we conclude that hsMSNA have considerable potential as drug delivery system carrier in the field of nanomedicine.
AcknowledgmentsWe are grateful to the National Key Basic Research Program of China (No. 20131970096) and the National Key R & D Plan (No. 2016YFC0304502).
| [1] |
C.T. Kresge, M.E. Leonowicz, W.J. Roth, et al., Nature 359 (1992) 710-712. DOI:10.1038/359710a0 |
| [2] |
S.F. Badylak, R.M. Nerem, Proc. Acad. Sci. India Sect. B 107 (2010) 3285-3286. |
| [3] |
B.T. Holland, C.F. Blandfor, T. Do, A. Stein, Chem. Mater. 11 (1999) 795-805. DOI:10.1021/cm980666g |
| [4] |
M. Grün, I. Lauer, K.K. Unger, Adv. Mater. 9 (1997) 254-257. DOI:10.1002/adma.19970090317 |
| [5] |
Q. Huo, J. Feng, F. Schüth, G.D. Stucky, Chem. Mater. 9 (1997) 14-17. DOI:10.1021/cm960464p |
| [6] |
T. Asefa, Z. Tao, Chem. Res. Toxicol. 25 (2012) 2265-2284. DOI:10.1021/tx300166u |
| [7] |
L. Qi, J. Ma, H. Cheng, Z. Zhao, Chem. Mater. 10 (1998) 1623-1626. DOI:10.1021/cm970811a |
| [8] |
M. Vallet-Regi, A. Rámila, R.P. del Real, J. Pérez-Pariente, Chem. Mater. 13 (2001) 308-311. DOI:10.1021/cm0011559 |
| [9] |
B.G. Trewyn, I.I. Slowing, S. Giri, et al., Acc. Chem. Res. 40 (2007) 846-853. DOI:10.1021/ar600032u |
| [10] |
G.V. Rama Rao, G.P. López, J. Bravo, et al., Adv. Mater. 14 (2002) 1301-1304. DOI:10.1002/1521-4095(20020916)14:18<1301::AID-ADMA1301>3.0.CO;2-T |
| [11] |
X.J. Wu, D. Xu, Adv. Mater. 22 (2010) 1516-1520. DOI:10.1002/adma.200903879 |
| [12] |
J. Liu, S.Z. Qiao, S.B. Hartono, G.Q.M. Lu, Angew. Chem. 122 (2010) 5101-5105. DOI:10.1002/ange.v122:29 |
| [13] |
J. Li, J. Liu, D. Wang, et al., Langmuir 26 (2010) 12267-12272. DOI:10.1021/la101225j |
| [14] |
Y.J. Wong, L. Zhu, W.S. Teo, et al., J. Am. Chem. Soc. 133 (2011) 11422-11425. DOI:10.1021/ja203316q |
| [15] |
Q. Yu, P. Wang, S. Hu, et al., Langmuir 27 (2011) 7185-7191. DOI:10.1021/la200719g |
| [16] |
L. Tan, D. Chen, H. Liu, F. Tang, Adv. Mater. 22 (2010) 4885-4889. DOI:10.1002/adma.201002277 |
| [17] |
H. Liu, D. Chen, L. Li, et al., Angew. Chem. Int. Ed. 50 (2011) 891-895. DOI:10.1002/anie.201002820 |
| [18] |
L. Li, F. Tang, H. Liu, et al., ACS Nano 5 (2011) 679. DOI:10.1021/nn103380r |
| [19] |
T. Suteewong, H. Sai, R. Hovden, et al., Science 340 (2013) 337-341. DOI:10.1126/science.1231391 |
| [20] |
F. Tang, L. Li, D. Chen, Adv. Mater. 24 (2012) 1504-1534. DOI:10.1002/adma.201104763 |
| [21] |
W. Feng, L. Chen, X. Zhou, et al., J. Control. Release 213 (2015) e108-e109. |
| [22] |
J. O'Connor, Y. Lang, J. Chao, et al., Small 10 (2014) 469-473. DOI:10.1002/smll.v10.3 |
| [23] |
S. Davidson, D.A. Lamprou, A.J. Urquhart, et al., ACS Biomater. Sci. Eng. 2 (2016) 1493-1503. DOI:10.1021/acsbiomaterials.6b00224 |
| [24] |
D.I. Meiron, G.R. Baker, S.A. Orszag, J. Fluid Mech. 114 (1982) 283-298. DOI:10.1017/S0022112082000159 |
| [25] |
O. Trofymluk, A.A. Levchenko, A. Navrotsky, Microporous Mesoporous Mater. 149 (2012) 119-125. DOI:10.1016/j.micromeso.2011.08.022 |
| [26] |
X. Yan, Z. Lei, J. Colloid Interface Sci. 362 (2011) 253-260. DOI:10.1016/j.jcis.2011.06.062 |
| [27] |
D. Zhao, J. Sun, Q. Li, G.D. Stucky, Chem. Mater. 12 (2000) 275-279. DOI:10.1021/cm9911363 |
| [28] |
N.A. Seaton, Chem. Eng. Sci. 46 (1991) 1895-1909. DOI:10.1016/0009-2509(91)80151-N |
| [29] |
M. Kruk, M. Jaroniec, A. Sayari, Adsorption 6 (2000) 47-51. DOI:10.1023/A:1008995015347 |
| [30] |
X. Wu, Z. Han, R.M. Schur, Z.R. Lu, ACS Biomater. Sci. Eng. 2 (2016) 501-507. DOI:10.1021/acsbiomaterials.5b00398 |
| [31] |
H.P. Jarvie, H. Al-Obaidi, S.M. King, et al., Environ. Sci. Tech. 43 (2009) 8622-8628. DOI:10.1021/es901399q |
| [32] |
Z.M. Cui, Z. Chen, C.Y. Cao, et al., Chem. Commun. 49 (2013) 2332-2334. DOI:10.1039/c3cc38649j |
| [33] |
P. Yang, S. Gai, J. Lin, Chem. Soc. Rev. 41 (2012) 3679-3698. DOI:10.1039/c2cs15308d |
| [34] |
Y. Song, Y. Li, Q. Xu, Z. Liu, Int. J. Nanomed. 12 (2016) 87-110. DOI:10.2147/IJN |
| [35] |
J.L. Vivero-Escoto, I.I. Slowing, B.G. Trewyn, V.S.Y. Lin, Small 6 (2010) 1952-1967. DOI:10.1002/smll.200901789 |
| [36] |
W. Xie, Z. Guo, F. Gao, et al., Theranostics 8 (2018) 3284-3307. DOI:10.7150/thno.25220 |
| [37] |
B. Cheng, H. He, T. Huang, et al., J. Biomed. Nanotechnol. 12 (2016) 435-449. DOI:10.1166/jbn.2016.2195 |
| [38] |
J. Peng, T. Qi, J. Liao, et al., Theranostics 4 (2014) 678-692. DOI:10.7150/thno.7869 |
| [39] |
M.Y. Kim, J. Kim, ACS Biomater. Sci. Eng. 3 (2017) 572-578. DOI:10.1021/acsbiomaterials.6b00716 |
| [40] |
K.M. Rao, S. Parambadath, A. Kumar, et al., ACS Biomater. Sci. Eng. 4 (2018) 175-183. DOI:10.1021/acsbiomaterials.7b00558 |
| [41] |
Q. Gao, W. Xie, Y. Wang, et al., RSC Adv. 8 (2018) 4321-4328. DOI:10.1039/C7RA12446E |
| [42] |
Q. Yang, J. Peng, Y. Xiao, et al., ACS Appl. Mater. Interfaces 10 (2018) 150-164. DOI:10.1021/acsami.7b14705 |