 2018, Vol. 29
2018, Vol. 29 


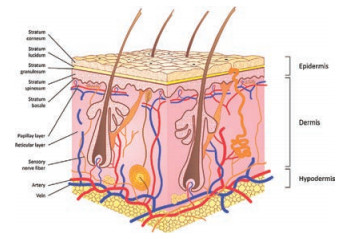
The skin is the largest and most visible organ of the body. The skin covers a total surface area of approximately 1.5–2 m2 and is the barrier between human body and the external environment [1]. Many of its functions include temperature regulation, immunity from microorganisms, maintaining electrolyte balance, as well as protection from physical injuries, chemical agents, and ultraviolet radiation [2, 3]. In addition, skin is also an important avenue for absorption of drugs and exerting their efficacy. The skin is composed of epidermis, dermis, and subcutaneous tissue, and contains appendages (such as hair follicles, sebaceous glands, sweat glands), blood vessels, lymphatic vessels, nerves, etc. The epidermis can be divided into five layers from the inside to the outside, namely the stratum basale, stratum spinosum, stratum granulosum, stratum lucidum, and stratum corneum (SC) (Fig. 1) [3, 4]. The properties of SC are quite different from those of the other layers, with the overall structure composed of inactive keratinocytes and intercellular lipids that form a 'brick and mortar' model, in which, the protein-rich keratinocytes serve as the bricks and the intercellular lipids serve as the mortar [5, 6]. SC is the main factor determining the skin barrier, and also the major obstacle limiting the rate of percutaneous absorption even though the thickness is only 10–20 μm [7, 8]. There are two routes of transdermal permeation of drugs. One is through the natural channel of skin appendages. These channels are hydrophilic and have a diameter of few microns. Owning to the fact that the average follicular orifice area on the human skin surface is only about 0.1% of the total surface area [9], it is not the primary pathway of percutaneous absorption. The second route is through the penetration of the epidermis, to enter the dermis through the SC and the deeper epidermis, being absorbed in the body circulation by the capillaries. As for the penetration of drugs and passing through the SC, two pathways exist, namely, the transcellular route, through which substances infiltrate the keratinocytes and intercellular lipids, and subsequently pass through and are transported. The drug needs to diffuse through hydrophilic and hydrophobic areas, and therefore, it may not be applicable to most drugs. The second and most likely route taken by drugs when penetrating the SC is via a tortuous pathway through the lipids surrounding the keratinocytes, known as the intercellular route (Fig. 2) [10].

|
Download:
|
| Fig. 1. Schematic illustration of the layers of the skin. Reproduced with permission [3]. Copyright 2015, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | |

|
Download:
|
| Fig. 2. The pathways for percutaneous absorption of drugs. | |
Transdermal drug delivery system (TDDS) refers to a route of drug delivery through the skin to achieve local or systemic therapeutic action. It is one of the focus areas of research for the third-generation pharmaceutical preparations, next only to oral medication and injection [11]. The reasons lie in the administration route of the drug, which is convenient, easy to use, non-invasive, and also improves patient compliance [12]. It also reduces the fluctuation of the drug concentration in the blood, provides steady plasma levels and fewer chances of overdose and easy detection of the drug [13, 14]. At the same time, it evades the gastrointestinal environment, such as pH, enzymatic activity, and the interference of drug and food interaction on the drug efficacy and the 'first pass effect' (where active drug molecules can be converted to inactive molecules or even to molecules responsible for side effects) by liver. These conditions lengthen the therapeutic effect of drugs with shorter half-life and enhance their long-term stability of drug [15]. Administration of drugs can be stopped at any point after removal of the stimulation from the site [16].
Although TDDS has many advantages, the use of drugs in TDDS is currently limited. As mentioned above, the most resistance during the percutaneous permeation of the drugs comes from the SC of the skin [17]. When many drugs are delivered through the skin, adequate permeability rate is difficult to achieve as per therapeutic requirements. To overcome these difficulties, nanotechnology may be a good choice. Nanotechnology refers to the technology of using a single atom or molecule to produce or process macromolecular matter into a material with a particle size of 1–100 nm. One of the important areas of nanotechnology is nano-formulations [18]. Given their small particle size, nanoformulations have a better effect on drug retention, specificity and targeting (Fig. 3) [19], which makes an ideal TDDS. They have many advantages, such as being painless, minimal skin injury (does not change the general structure of SC of the skin and does not destroy the skin barrier function), and promotes permeation of macromolecular drugs, which has become a very popular field of research on TDDS [20]. Nano-formulations can be divided into vesicles including liposomes, transfersomes, ethosomes, niosomes, invasomes, and nanoparticles including lipid nanoparticles, polymeric nanoparticles and nano-emulsions (Fig. 4). As for active transdermal administration, microneedles are not involved, instead, ultrasonic, electroporation, hot perforation and comprehensive application of other methods enhancing penetration are used. This review is based on the advances in research on nano-formulations of passive TDDS, and focuses on the classification, components, characteristics, transdermal mechanism and application of nanoformulations (Table 1). We hope to present a foundation for future research in nano-formulations for TDDS, and to enhance our understanding for clinical and therapeutic applications.

|
Download:
|
| Fig. 3. A schematic representation highlighting the benefits of drug-delivery using nano-formulations. | |

|
Download:
|
| Fig. 4. The structures of nano-formulations. | |
|
|
Table 1 Summary of typical components, transdermal mechanism and applications of nano-formulations. |
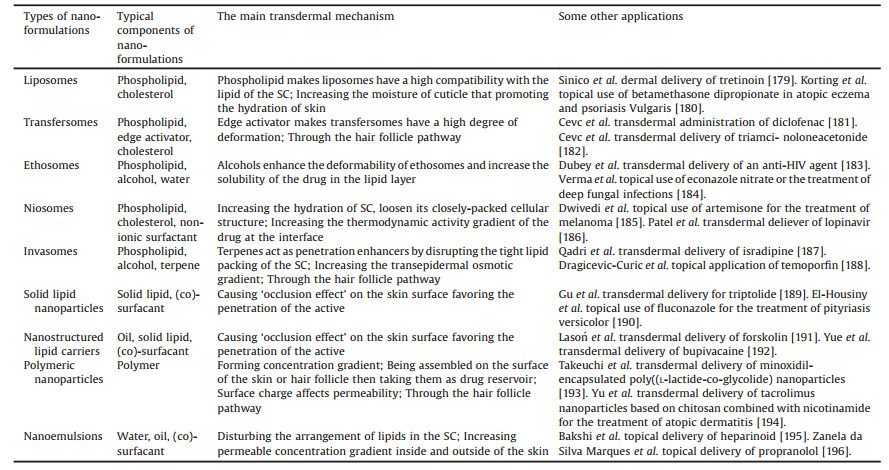
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2. Classification and application of nano-formulations 2.1. Vesicles
Vesicles are water-filled colloidal particles. The walls of these capsules consist of amphiphilic molecules in a bilayer conformation [21]. In conditions of excess water, these amphiphilic molecules form one (unilamellar vesicles) or more (multilamellar vesicles) concentric bilayers [22]. With the goal of increasing the penetration function of the components, vesicles can carry watersoluble and liposoluble drugs to achieve transdermal absorption. When applied to topical application, vesicles can be used for drug storage to achieve sustained release. With respect to TDDS, they can regulate the absorption rate through the multilayer structure [23]. Due to the presence of different components, the vesicular system can be divided into several types. Liposomes are mostly made up of phospholipids and were discovered by Bangham in 1960 [24], but liposomes first used for topical treatment were reported by Mezei and Gulasekharam [25]. Non-ionic surfactantbased vesicles discovered by L'Oreal in the 1970s are known as niosomes [26]. Niosomes are formed of non-ionic amphiphiles in aqueous media resulting in closed bilayer structures [27]. Cevc and Blume introduced the first generation of the highly deformable, elastic liposomes in 1992, and transfersomes appeared soon [28]. Subsequently, the second generation of elastic vesicles, mainly consisting of non-ionic surfactants, was introduced in 1999 by van den Bergh [29]. In 1997, Ethosomes, the new soft vesicular carriers mostly consisting of phospholipids and ethanol, were developed by Touitou et al. [30] Invasomes were developed by Fahr et al. in 2004, and they were composed of phosphatidylcholine, ethanol and a mixture of terpenes [31]
2.1.1. LiposomesLiposomes are circular soft-matter vesicles formed from one or more bilayer membrane(s) separating an aqueous medium from another. Their major components are usually phospholipids with or without cholesterol. Phospholipid molecules are mainly composed of different polar head groups and two hydrophobic hydrocarbon chains. The polar groups can be zwitterionic or negatively charged. The hydrocarbon chain molecules have different lengths and possess different degrees of unsaturation. The formation of liposomes occurs spontaneously upon reconstitution of dry lipid films in an aqueous solution [32]. This unique structure allows liposomes to have both hydrophilicity and hydrophobicity to encapsulate both water-soluble and liposoluble substances [33]. In 1980, Mezei and Gulasekharam first reported the use of liposomes as topical drug delivery carriers [32]. Bangham created liposomes containing triamcinolone acetonide that had 4–5 times higher concentration than the normal liposomes in rabbit epidermis and dermis, lower concentration in the thalamic region (a possible site of adverse effect), and reduced urinary excretion when compared with that of the control [24]. Some studies however, show that liposomes can only remain on the surface of the skin, and cannot penetrate the stratum granulosum of epidermis, which leads to minimal amounts of drug absorbed by the circulating blood. They are able to only increase the retention of drugs that stay in the skin, prolong their activity at the site of the lesion, and enable long-term sustained release. Therefore, it is mostly used for topical treatment of skin diseases [34], the details are discussed in this review.
Ning et al. prepared clotrimazole-loaded multilamellar liposomes in order to develop alternative formulation for the vaginal administration of clotrimazole to provide sustained and controlled release of appropriate drugs for local vaginal therapy. The in vitro permeation data were that the liposomes increased the clotrimazole total penetration through the vaginal mucosa, and the accumulation of clotrimazole in the mucosa also increased as compared to the control group during the 24 h period [35]. Seong et al. prepared surfactant-stable and pH-sensitive liposomes coated with N-succinyl chitosan (NSC) and chitosan oligosaccharide (COS) for delivery of quercetin. A four-layered liposome was successfully prepared using COS and NSC. They compared the permeability of the four-layered liposome with those of the uncoated liposomes, and a sample in which quercetin was dissolved in the chemical 1,3-butylene glycol (1,3-BG). The cumulative permeation amount of quercetin after 24 h was in the order of the four-layered liposome (21.10 μmol/L) > uncoated liposomes (16.08 μmol/L) > 1,3-BG (9.49 μmol/L), and the amount of quercetin released was highest for the four-layered liposome. The results showed that liposomes improved the drug release and skin penetration compared to the normal solution, suggesting the possibility of developing a transdermal delivery system for effectively delivering poorly soluble drugs [36]. Allantoin is traditionally employed in the treatment of skin ulcers, but it has very low skin penetration ability due to its low logP ~3.14 [37]. Manca et al. incorporated allantoin in the liposomes and alternatively in liposomes enriched with arganoil (ARG liposomes) to prolong the residence time at the action site and improve the local accumulation. In this work, allantoin in water dispersion and gel formulation (Sameplast® gel: a commercial product containing allantoin 1%, dispersed in a gel of hydroxy ethylcellulose and xanthan gum prepared in water and glycerol), were used as a comparison. The accumulation of conventional liposomes loaded with allantoin in the dermis doubled (-3.4 g/cm2, P < 0.05) with respect to that provided by Sameplast® gel. When the drug was entrapped in the ARG liposomes, they seem to provide a softening and relaxing effect on the skin. An important improvement in drug accumulation was observed in the epidermis (-2.2 g/cm2, P < 0.05 versus all), and mostly in the dermis (-8.7 g/cm2, P < 0.01 versus all). Additionally, liposomes and ARG liposomes, significantly increased the amount of drug permeated through the skin and recovered in the receptor fluid, which was -33 g/cm2 (P < 0.05 versus dispersion and Sameplast® values) but it was significantly lower (-17 g/cm2) when water dispersion or Sameplast® gel were used. Above all, we now understand the distinct advantages of liposomes for TDDS, such as for improving drug release, drug accumulation, and skin penetration. Further modifications to the composition could greatly improve their therapeutic potential [38].
2.1.2. TransfersomesThe term Transfersomes®, was proposed by Cevc et al., which are also called deformable liposomes or elastic or ultra-flexible liposomes [39]. The most important feature of the vesicles is the elasticity caused by adding a single-chain surfactant (also described as the edge activator) such as sodium cholate, Tween®, Span®, polysorbic acid, and dipotassium glycyrrhizinate. These surfactants weaken the phospholipid bilayer and render the vesicles with ultra-deformability, and these became the first generation Transfersomes® [21, 40]. Over time, the secondgeneration Transfersomes® made of at least one basic bilayer builder, typically phosphatidylcholine with fluid-chains, and at least two more polar lipophilic substances (such as one surfactant and one surfactant mimicking drug) emerged [41]. The thirdgeneration of Transfersomes® are combinations of amphiphilic surfactants with or without phospholipids [42]. The discovery of transfersomes, due to their deformability, as they are able to pass through skin pores 5–10 times lesser than their own size, made the relative molecular mass limit of the drugs that penetrate the skin reach up to 1 million. It also enabled administering macromolecular drugs such as peptides or proteins through TDDS [43].
Zheng et al. used transfersomes loaded with itraconazole, used as a model drug to compare their permeation with regular liposomes on rat skin. They showed that the permeation amount of transfersomes was small at the start of the test, but in 2 h the transmission of transfersomes began showing marked improvement, and so with time. The permeation amount accumulated in 48 h was up to 90%, which was about 2.2 times more than that of regular liposomes [44]. Duangjit et al. prepared meloxicam-loaded cationic transfersomes, and the cationic transfersomes provided greater meloxicam skin permeation than the conventional liposomes and meloxicam suspensions [45]. Choi et al. made red ginseng employing a new vesicular system of transfersomes to improve the topical delivery of ginsenoside Rhl isolated from red ginseng. They showed that the transfersomes provided a significantly higher skin permeation of ginsenoside Rhl compared to the conventional liposomes on the rat dorsal skin [46]. Sildenafil citrate (SLD), a selective cyclic guanosine monophosphate-specific phosphodiesterase type 5 inhibitor used for the oral treatment of erectile dysfunction, and more recently, for other conditions, including pulmonary hypertension [47]. The challenges of oral administration of the drug including poor bioavailability and short duration of drug activity requires frequent administration [48]. Thus, Badr-Eldin et al. formulated optimized SLD nanotransfersomal transdermal films with enhanced and controlled permeation aiming at surmounting the previously mentioned challenges, and hence improving the drug bioavailability. The optimized transfersomal films showed a controlled gradual release over the study period reaching a maximum amount of drug permeated of 1.54 folds compared with the control films. In addition, the computed permeability and diffusion coefficients for the optimized films were 1.25 and 1.57 folds higher in comparison to the control films. The mean SLD plasma concentrations confirmed that the optimized transfersomal films were more stable than the other films [49]. There are also reports about ketoconazole [50], resveratrol [51], and eprosartan mesylate (EM) [52] loaded with transfersomes. Based on the literature, transfersomes have the ability not only to penetrate the SC barrier and reach deeper dermal tissues but also in the systemic circulation owing to their flexibility and deformable structure.
2.1.3. EthosomesEthosomes were first developed by Touitou in 1996, and reported in 2000 [53]. Ethosomes are composed of phospholipids, alcohol and water. Compared to the liposomes, ethosomes are characterized by their high concentration of alcohol. Ethosomes promote percutaneous permeation of drugs and, besides, the phospholipid also contributes to it. As the molecule of water near the head of the lipid bilayer is replaced by alcohol, it increases the flexibility and fluidity of ethosomes. Ethosomes have the characteristic size of a small particle, stable structure and high entrapment efficiency that can delay the release of drugs [54], therefore, compared with the ordinary liposomes, the ethosomes can carry the drug to the skin much deeper or directly into the blood circulation system and significantly improve the drug transdermal efficacy [53, 55]. Compared to the transfersomes, the ethosomes are a type of multiphase dispersion system, with better stability and longer retention periods [56]. Other excipients usually added in the ethosomes formulation include cholesterol (vesicle membrane stabilization) [57], rhodamine (marker dyes for characterization study) [58], carbopol [59], pluronic F 127 [60], and poloxamer 407 (gel-formers used to produce vesicular gels in order to increase residence time) [61].
Diabetes is a global public health problem threatening human health and is becoming an important cause of prolonged ill health and early death [62]. Siddhodhan et al. studied the usefulness of ethosomes for transdermal delivery of repaglinide (RPG) in the treatment of type Ⅱ diabetes mellitus in rats. The ethosomes loaded with RPG were prepared from dipalmitoyl phosphatidylcholine and ethanol by the cold method. RPG ethosomes possessing the size of 0.171–1.727 mm and entrapment efficiency of 75%–92% were obtained. They demonstrated a significantly higher permeation (64%–97% of the administered dose) across excised rat skin than the free drug and its hydro-alcoholic solution. The blood glucose level in rats that received oral RPG drastically (P < 0.01) reduced to 98.7 ± 11.7 mg/dl over 11 h. Thereafter, hypoglycemic activity due to RPG was not noticed as blood glucose levels raised up to 209 ± 7 mg/dl by the end of 24 h, probably owing to its short half-life. However, a prolonged antidiabetic drug activity was displayed by the ethosomal RPG, suggesting a sustained release from the transdermal formulation. Also, the RPG ethosomal gel-treated rats elicited significantly lower (P < 0.01) blood glucose level (97.3 ± 3.5 mg/dl) at the end of 24 h. The study demonstrated that with the help of transdermal drug delivery of ethosomal system, it is possible to prolong drug release to reduce the dosage frequency for the treatment of type Ⅱ diabetes mellitus [63]. Li et al. made lornoxicam ethosomes gels with enhancers such as azone, menthol, and lauryl alcohol, which are used for in vitro transdermal delivery experiments to investigate skin permeability supplemented with penetration enhancers, and evaluate the in vivo pharmacodynamics of these formulations. The results showed that compared to other formulations, the optimized lornoxicam ethosomal gels with 5% menthol significantly increased transdermal penetration. Meanwhile, the optimized lornoxicam ethosomal gels showed remarkable anti-nociceptive and anti-inflammatory activity compared to plain lornoxicam gels [64]. These results suggest that the ethosomes are promising drug carriers for TDDS to enhance drug transdermal efficiency and therapeutic effect.
2.1.4. NiosomesNiosomes, also called non-ionic surfactant-based elastic vesicles, are molecular clusters formed by self-association of non-ionic surfactants in an aqueous phase, in addition to, in many cases, cholesterol or its derivatives [65]. They are the second-generation elastic vesicles that can be divided into three categories depending on their size, as: small unilamellar vesicles (SUV) (10–100 nm), large unilamellar vesicles (LUV) (100–3000 nm), and multi-lamellar vesicles (MLV), in which more than one bilayer is present [66]. Without consideration of the size, niosomes including proniosomes [67], surfactant ethosomes [68], elastic niosomes [69], polyhedral niosomes [70], discomes (disk-shaped vesicle) [71], aspasomes (ascorbyl palmitate vesicle) [72] and so on. Compared to liposomes, the carrier material of niosomes does not contain phospholipids, whichavoidsthe oxidative degradation of phospholipids leading to a high chemical stability. There are many types of surfactants easily derivatized and offering higher versatility to the niosomes, no special conditions are needed for their production and storage, and the process is simplified to achieve large-scale production with a lower cost [73]. Niosomes are osmotically active which could improve the solubility of some poorly soluble drugs and bioavailability. Furthermore, entrapping of lipophilic drugs into vesicular bilayer membranes and hydrophilic drugs in aqueous compartments, ease of handling, availability of prepared materials in pure form are some of the advantages of niosomes [74].
Dyslipidemia is one of the most important risk factors for many chronic non-communicable diseases resulting in serious morbidity, mortality, and medical costs worldwide [75]. The prevalence of fibrous plaques considerably increases from 8% to 69% from childhood to young adulthood, which highlights the fact that the development of atherosclerotic cardiovascular disease begins early in life, as early as in childhood [76]. Statins are one of the first-line medications used in the pediatric population with dyslipidemia [77]. Zidan et al. prepared simvastatin-loaded niosomal gels to improve their hypolipidemic efficacy. The in vivo pharmacokinetic investigations in rats showed an augmentation in simvastatin bioavailability from its transdermal niosomal formulations by about 3 folds compared with oral drug suspension. The results were well supported by investigating the corresponding hypolipidemic effects to show a significant enhancement of the biological activities. So, simvastatin niosomal gels could be considered promising for TDDS, particularly for the treatment of hyperlipidemic pediatric patients [78]. Capsaicin is the pungent principle of red pepper used in topical therapy for a variety of disorders like pain and inflammatory diseases [79]. However, the strong pungency of capsaicin leads to its limited clinical use, a significant first pass metabolism has been detected from capsaicinoids in rats, and the half-life of capsaicin by intravenous administration in rats was very short (7.06 min) [80]. Lorena et al. performed a new study on niosomal formulations obtained from a mixture of commercial surfactants to be applied topically for the release of capsaicin. Niosomes were compared to microemulsions prepared from the same surfactants in the same ratio to evaluate their possible use in TDDS. The results revealed a lower amount of capsaicin that permeated from microemulsions in 12 h when compared to niosomal formulations [26]. These results indicate that niosomes may be a favorable drug delivery system, and superior to some other vesicular carriers.
2.1.5. InvasomesInvasomes, introduced by the Verma and Fahr groups [81], are vesicles composed of phosphatidylcholine (soy-phosphatidylcholine, lysophosphatidyl choline), ethanol and a mixture of terpenes. Soy-phosphatidylcholine forms the invasomal bilayer matrix. Lysophosphatidyl choline acts as an edge activator giving flexibility to the phosphatidylcholine bilayers. Both ethanol and terpenes act as penetration enhancers for drugs and also endow fluidity or flexibility with the phospholipid bilayers [82]. Invasomes were shown to be efficient drug delivery systems for both hydrophilic and lipophilic agents [81, 83].
Isradipine is an effective calcium channel blocker used in the management of hypertension. It undergoes extensive first pass metabolism and has low oral bioavailability. Hence, Qadri et al. attempted to prepare and characterize invasomes carrier for isradipine. The optimized formulation produced a particle size of 194 ± 18 nm, entrapment efficiency (88.46%), and attained a mean transdermal flux of 22.80 ± 2.10 mg cm-2 h-1 through rat skin. Confocal laser scanning microscopy revealed an enhanced permeation of rhodamine-red-loaded isradipine invasomes to the deeper layers of the rat skin. In an anti-hypertensive study, isradipine invasomes formulation led to a 20% reduction in blood pressure by virtue of better permeation through the skin of Wistar rats, with substantial and constant decrease in blood pressure, for up to 24 h [84]. Kamran et al. formulated, optimized and evaluated the transdermal potential of novel vesicular nano-invasomes, containing anti-hypertensive agent. They compared BENICAR® (a marketed tablet formulation of olmesartan) and an invasomal gel containing olmesartan in a rat model. The optimized formulation was further evaluated for in vitro drug release and in vivo pharmacokinetics. The optimum nano-invasomes formulation had vesicle size of 83.35 ± 3.25 nm, entrapment efficiency of 65.21% ± 2.25%, and transdermal flux of 32.78 ± 0.703 (μg cm-2 h-1). The pharmacokinetic study revealed that the formulation containing transdermal nano-invasomes had 1.15 times improvement in bioavailability of olmesartan with respect to the control formulation in Wistar rats. The plasma concentration profile of olmesartan following the application of optimized invasomal gel showed that the Cmax of olmesartan gel was significantly (P < 0.05) lower than after application of the oral tablets, and the AUC values were significantly higher indicating increased bioavailability as well as longer duration of activity via the transdermal route of administration by invasomal gel [85]. So, invasomes could be a useful carrier system for transdermal delivery of many drugs.
2.2. NanoparticlesThe terms 'nanomaterials' or 'nanoscale' are used to describe objects with a size in the range of 1–100 nm [86, 87]. Nonetheless, particles with sizes ranging from 1 nm to 1000 nm have been approved as nanocarriers [88]. So, in this review, the term nanoparticles will be used for particles with a size in the range of 1–1000 nm, and used for TDDS. The drugs can be encapsulated or dissolved in the nanoparticles and can also be adsorbed or coupled on their surface [89]. After the drug is administered in the form of nanoparticles, it can offer drugs with targeted and controlled release effects, change the dynamics of drugs in vivo, prolong the residence time in the blood, thereby improving the bioavailability of drugs, reducing the toxic and side effects, and improved drug efficacy [90]. Nanoparticles are usually formed by polymerization and cross-linking, and biodegradable polymer materials, such as gelatin and polylactic acid are often used [91]. Additional studies on lipid nanocarriers and polymeric nanoparticles are ongoing, and will be discussed in this review.
2.2.1. Lipid nanocarriersLipid nanocarriers are a class of colloidal drug delivery systems with lipid as the carrier or material. The system was established in 1990s [92], liposomes are the early lipid nanocarriers. The advanced and more complex lipid nanocarriers, such as solid lipid nanoparticles (SLN), nanostructured lipid carriers (NLC), and some polymeric nanoparticles composed of a blend of lipids have emerged [93]. When the lipid nanoparticles adhere to the skin surface, the particles are stacked, which fuse and deform due to capillary force, forming a membrane on the skin surface. This membrane reduces the moisture loss on the skin surface and increases the hydration of the skin, thus promoting the permeation of the skin. Besides, the lipid nanocarriers improve the stability of the drug, and the encapsulation effect [94].
SLN were the first-generation of lipid nanoparticle drug delivery system developed after microemulsions, liposomes and polymeric nanoparticles [95]. They are characterized by the presence of a mixture of lipid or blend lipids with good physiological compatibility and remain as solids at room or body temperature. They encapsulate the drug in the lipid core or adsorb on the surface of the particle with an average diameter below 1000 nm [96]. Other common excipients used in SLN formulations are emulsifiers, co-emulsifiers, and water. SLN not only is safe like the conventional liposomes, but also has similar stability and biocompatibility like the polymeric nanoparticles. Furthermore, the introduction of high pressure homogenization technology in the preparation of SLN facilitates mass production [97].
Zhang et al. formulated SLN as an oil-in-water microemulsion for transdermal delivery of aconitine to improve its safety and permeability. Drug encapsulation efficiencies for these formulations were higher than 85%, and compared with an ethanol tincture, the tested solid-lipid nanoparticle formulations achieved improved transdermal fluxes and drug deposition in skin in vitro. In vivo microdialysis of aconitine in rat dermis resulted in markedly higher concentration following application of SLN compared to tincture throughout the experimental period. The superior transdermal permeability of aconitine-loaded SLN contributed to stronger anti-inflammatory and analgesic effects on mouse in vivo models of pain than the tincture (P < 0.05) [98]. Psoriasis is a common, chronic, immune-mediated disease, affecting over 125 million people around the world, associated with significant negative impact on the patient's quality of life [99]. In psoriasis, the inflammatory process is strongly related to an excessive immune response, thus immuno-suppressants, such as methotrexate (MTX) have shown good efficacy [100]. Due to the variable intestinal absorption as well as first-pass metabolism in the liver, orally-administrated MTX has a relative lower bioavailability to the one obtained by parenteral administration [101]. Etanercept is one of the FDA-approved existing TNF-α inhibitor molecules for moderate to severe plaque psoriasis treatment [102]. In view of the individual contribution of MTX and etanercept in the treatment of psoriasis, it is possible to envisage a combination therapy into a targeted approach. So, Ferreira et al. investigated SLN as delivery systems for MTX and etanercept combination therapy and to develop a simple and unique hydrophilic formulation for topical psoriasis treatment. MTX was loaded into SLN and etanercept conjugated SLN by hot ultrasonication method. Both types of SLN were further incorporated into a carbopol hydrogel for easy topical application. The in vitro skin permeation was evaluated in pig ear skin as a model, and human biopsies from healthy and psoriatic skin to determine the potential skin deposition and permeation. After 8 h, 1.7% ± 0.1%, 3.1% ± 0.1% and 3.8% ± 0.2% of MTX permeated the skin delivery as free, loaded in protein-conjugated SLN, and loaded in SLN, respectively. At the end of the permeation assay, about 38% of the initial added amount of MTX did not penetrate or permeate the skin, while 22% and 15% were found for the conjugated-SLN and SLN formulations, respectively. Incorporating MTX in the lipid nanoparticles led to a significantly higher accumulation of MTX in the skin compared to its free solution [103].
Compared with conventional liposomes and emulsions, SLN regulates the drug release rate and has a remarkable impact on hydration of skin, but the limited drug loading is a disadvantage. The solid lipids easily undergo polymorphic transition to decrease formulation stability, which induces drug expulsion from SLN. To overcome the limitation, the second-generation lipid nanoparticles-NLCs were developed [104]. The difference between NLC and SLN is the inner structure. Besides solid lipids, the structure of NLC contains a certain amount, usually up to 30% of an oil [105]. When the NLC adheres to the skin surface, they augment skin hydration and promote the deposition of drugs by reducing the keratinocytes packing and widening the inter-keratinocytes gaps [106]. So, compared to SLN, NLC increases drug encapsulation, drug-loading rate, and reduces the leakage of drugs in storage. The chemical similarity to skin lipids, the existence of a solid matrix, the highly specific surface area linked to their nanometer-scale size, and the biocompatibility makes them suitable for long-term skin administration [107].
Basal-cell carcinoma (BCC) is the most common form of skin cancer in the white population, with the incidence rate increasing at over 4% annually in all countries and social classes. It has been estimated that approximately 50% of all fair-skinned individuals (40–60 years old) are developing this type of skin cancer [108]. For treating such cutaneous malignant skin lesions, topical photodynamic therapy (PDT) is the latest and extremely beneficial option, which involves the use of a photosensitizing agent [109]. The second-generation photosensitizer, 5-aminolaevulinic acid (5-ALA), is a potential drug candidate as it is the precursor of the potent endogenous photosensitizer protoporphyrin Ⅸ (PpⅨ) [110]. The major problem associated with the conventional topical formulation of 5-ALA for PDT is insufficient drug penetration into the skin due to its hydrophilic nature. Qidwai et al. developed and optimized a topical NLC formulation of 5-ALA to enhance its penetration in the skin to the basal cells layer, thereby enhancing its cytotoxic effect to treat BCC. They found that 5-ALA NLC exhibited a controlled drug release, with 54% of the drug released within 5 h. In contrast, the drug release from the drug solution showed rapid release of 80% drug in 5 h. The NLC showed approximately 4 times higher cytotoxicity than the drug solution, which confirms the higher cellular uptake of NLC and considerably reduced cell viability via NLC formulation in contrast to the drug solution. NLC is a promising drug carrier to increase the accumulation of PpIX by enhancing the penetration of hydrophilic drugs, such as 5-ALA into deep skin layers, and improving the efficacy of topical PDT [111]. Babaei et al. compared the encapsulation of lidocaine-loaded NLC and nanoethosomes for improving its dermal delivery, and consequently, the local anesthetic efficacy. Lidocaine-loaded NLC and nanoethosomes were characterized by various techniques and used for an in vitro skin penetration study using excised rat skin and Franz diffusion cells. Optimized lidocaine-loaded NLC (encapsulation efficiency 69.86% and loading capacity 10.47%) and nanoethosomes (encapsulation efficiency 40.14% and loading capacity 8.02%) were chosen for a skin drug delivery study. The result showed that higher skin drug deposition of NLC and nanoethosomal formulations compared to lidocaine hydroalcoholic solution represented a better localization of the drug in the skin. NLC formulation showed the lowest entered drug in the receptor phase of Franz diffusion cell in comparison with nanoethosomes and hydroalcoholic solution confirming the highest skin accumulation of the drug. Both colloidal systems showed superiority over the drug solution for dermal delivery of lidocaine. NLC exhibited even more promising characteristics than nanoethosomes regarding drug loading and skin targeted delivery [112]. Pioglitazone (PZ) is used in the treatment of type Ⅱ diabetes [113]. PZ (BCS class Ⅱ) is a waterinsoluble drug with a short biological half-life of 3–5 h and is eliminated rapidly. It is rapidly absorbed and extensively metabolized by hydroxylation and oxidation to active and inactive metabolites in the liver [114]. Alam et al. design PZ-loaded NLC to investigate the bioavailability improvement by transdermal delivery. The in vivo pharmacokinetic study showed 2.17 times higher bioavailability, while the pharmacodynamics study showed that PZ NLC-based transdermal therapeutic system lowered the blood sugar level in a sustained pattern for a prolonged period of time as compared to Piosys tablet (marketed). These results clearly provide a possibility that the above NLC-based TTDS is a potential controlled release formulation for PZ, and could be a promising drug delivery system for the treatment of diabetes [115].
2.2.2. Polymeric nanoparticles (NPs)Polymeric NP are solid colloidal carriers prepared from the polymeric matrix similar to synthetic, semi-synthetic or natural polymers with size ranging from 10 nm to 1000 nm [116]. In the field of TDDS, the polymeric NP have been attracting more attention due to the fact that they can overcome the limitations of other lipid systems, such as they offer protection to the unstable drugs from degradation and denaturation, release continuously to reduce the side effects of toxic drugs, and increase the concentration gradient to enhance the percutaneous permeation of drugs [117]. Based on their method of preparation and architecture, the polymeric NP can be classified into nanospheres, nanocapsules and polymeric micelles [118]. The polymers widely used are synthetic polyesters such as poly(lactic acid) (PLA), poly((DL-lactide coglycolide) (PLGA), polycaprolactones (PCL), polyacrylates (PCA), poly (ε-caprolactone), polyacrylic acid (PAA), polyalkyl cyanoacrylate, poly (methyl methacrylates), and natural polyesters chitosan, gelatin, alginate or other polymer carriers [119]. These polymeric chains are made up of two or more homopolymer units covalently bound and can be synthesized into di-, tri- or multiblock, random, star and graft. Under some conditions they can form complex structures such as synthetic membranes that mimic the lipid bilayer of the cell membrane, but due to high molecular weight polymer chains, the polymer membrane structure is highly entangled, which is the biggest difference compared to the lipid bilayer [120]. Due to this reason, polymeric NP characterized by high mechanical strength, and non-deformability could not pass through pores smaller than or equal to their own size. Otherwise, it can be difficult for them to self-degrade, which means that they could retain drugs for longer periods of time than releasing them from nanoparticles and diffusing them into deep layers of skin [121].
Sunburn is a common recognition caused by the ultraviolet radiation (UVA radiations (320–400 nm) passing through the ozone layer and lead to premature aging of the skin by suppressing the immune function. UVB radiations (290–320 nm) are blocked by ozone layer to some extent, but still may be responsible for sunburn exposure [122], especially in fair-skinned individuals. The hyperalgesia caused due to repeated sunburn is associated with accelerating aging and skin cancer [123]. To reduce exposure to the UVR, one of the major practices prescribed by –The Skin Cancer Foundation, The American Academy of Dermatology, and The American Cancer Society is the use of sunscreen products [124]. Shetty et al. developed novel sunscreen creams containing polymeric NP of morin (an important plant flavonoid possessing both antioxidant and UVR protection properties) to optimize the delivery of the material into the skin. The amount of morin permeated from the nanoparticulate suspension at the end of 12 h was much higher than that observed with the morin plain suspension. This may be due to the increased ability of polymeric NP to penetrate the skin. Besides improving drug permeation, the polymeric NP resulted in higher skin deposition of morin indicating that the particles are also being retained to a larger extent within the skin, in addition to resulting in higher skin permeation of morin (skin retention of morin was 10.71 ± 0.98 μg/cm2 and 35.79 ± 1.27 μg/cm2 from plain morin suspension and morin polymeric NP, respectively). We know that the polymeric NP yielded good skin permeation as well as retention for morin [125]. Batheja et al. successfully synthesized chitosan-modified PLGA nanogel and modified it with oleic acid. The results showed that the PLGA nanogel could significantly improve the skin permeability of diclofenac [126]. Other polymeric systems like micelles and dendrimers have been used in TDDS. Compared to other delivery vehicles, their inability to effectively cross the skin barrier via any other route apart from the shunt pathway drastically reduces their efficacy. As research on these is relatively limited [127, 128], they are not discussed in this review.
2.3. Nanoemulsions (NE)NE are low viscosity, isotropic, thermo-dynamically and kinetically stable (without any apparent flocculation or coalescence during long-term storage) mixtures. The mixture consists of transparent or translucent oil globules dispersed in aqueous phase stabilized by an interfacial film of surfactant or co-surfactant molecules of extremely small droplet size (the mean droplet diameter attained is usually less than 500nm) [129]. As for the particle size of NE, which has been debated; the upper particle size limits for NE of 1000nm, 500nm, 200nm, and 100nm have been proposed [130, 131]. The word NE should not be confused with microemulsions. Despite NE having the nearly same droplet size range, composition, and appearance as microemulsions, they differ tremendously in structural aspects and long-term thermodynamic stability [132]. The small particle size, large specific surface area and low surface tension of NE give them good wettability to retain them in close contact with the skin. Besides, NE offers many other advantages including higher solubilization capacity, physical stability, improved bioavailability, ease of preparation, produced with less energy input, and a longer shelf life. Compared to commonly used topical skin preparations, the transdermal time of NE is shorter and the effect of percutaneous absorption is better [133]. Based on the components, NE can be classified into three types i.e., oil in water (O/W: oil phase dispersed in continuous aqueous phase), water in oil (W/O: water phase dispersed in continuous oil phase), bi-continuous/multiple emulsion (micro domains of oil and water phases are inter-dispersed within the system) [134].
Reports reveal that O/W NE is being increasingly used as a delivery system for encapsulating lipophilic components in pharmaceutical products. Kwasigroch et al. prepared a multifunctional O/W NE with a mean droplet diameter about 200nm and a low polydispersityindex (< 0.2) loaded with an anti-inflammatorydrug (indomethacin). The in vivo anti-inflammatory effect of the indomethacin alone or that loaded into the NE was measured by the carrageenan-induced paw edema in rats. The plain NE was shown to be innocuous in cellular studies and had no acute toxicity (observed in a rat model). NE loaded with indomethacin presented a significantly different anti-inflammatory response than the free drug. The edema of the groups that received indomethacin (2mg/mL) alone or dispersed in the NE increased to 55.4% ± 7.4% and 42.4% ± 17.1%, respectively. So, NE has great potential for enabling new advances in pharmaceutical science, especially with regard to TDDS applications [135]. In order to achieve deep skin delivery for tackling psoriasis, NE loaded gel containing clobetasol propionate and calcipotriol has been evaluated by Kaur et al.Intheir study, imiquimod-induced psoriatic BALB/c mice revealed significantly higher anti-psoriatic activity of NE gel as compared to free drugs and marketed formulation. The formulation developed had negligible skin irritation despite increased penetration into theskin [136]. Caffeine has been investigated for the treatment of various types of cancers by the oral administration route.There is also some evidence that dermallyapplied caffeine protects the skin from skin cancer caused by sun exposure. Shakeel et al. dispersed caffeine in an aqueous solution and titrated it against lauroglycol 90, transcutol HP and isopropanol to arrive at a thermodynamically stable and safe NE which was sized between 20–100nm. The in vitro skin permeation studies were performed on Franz diffusion cells using rat skin as the permeation membrane. Upon testing NE against aqueouscaffeine, they observed significant increase in permeability parameters, steady-state flux and permeability coefficient, which suggests that NE are good carriers for transdermal delivery of caffeine [137]. Chalcones are compounds of low water solubility that have been described as promising molecules for the treatment of cutaneous leishmaniasis (CL). A new chalcone derivative, (E)-3-(3-nitrophenyl)-1-(3, 4, 5-trimethoxyphenyl)prop-2-en-1-one (called SC) was synthesized by de Mattos et al. who reported promising activity against both promastigotes and intracellular amastigotes of Leishmania amazonensis. SC was successfully incorporated in NE using a spontaneous emulsification method. The result showed NE with high activity against intracellular amastigotes of L.amazonensisin THP-1 cells.Furthermore, when the parasitic inhibition profile is considered, the NE was both stabile and maintained leishmanicidal activity [138].
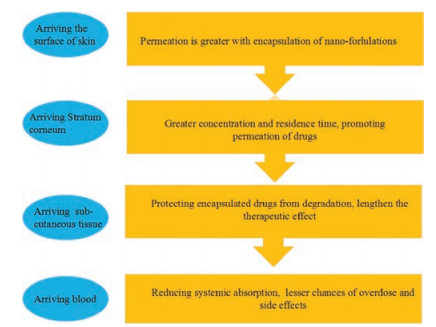
3. Transdermal mechanism of nano-formulationsDepending on the physical characteristics of their inherent small particle size and large specific surface area, nano-formulations can react fully with the skin surface and promote drug transdermal absorption. However, different nano-formulations interact with the skin through different mechanisms. For example, lipid-based nano-formulations have structural similarities with those comprising the epidermis and in particular the SC. So, they could attach onto the skin surface and increase skin hydration, gradually leading to a loose structure, polarity alteration, fluidization and even lipid exchange within the intercellular lipid domain (Fig. 5). Therefore, it is necessary to have a clear understanding of the transdermal mechanism of nano-formulations that can help us achieve better penetration and absorption of the drugs loaded with nano-formulations into the skin.

|
Download:
|
| Fig. 5. Interaction mechanisms of lipid-based nano-formulations with corneocytes. | |
{kind=link}
3.1. Liposomes
We previously discussed some studies that have shown that liposomes can only remain on the surface of the skin, but another report offers contrasting evidence [139]. This is because the application to dissimilar skins (animal or human) via diverse protocols may reveal different mechanisms, and liposomes depending on the composition and method of preparation, can vary with respect to size, lamellarity, charge, membrane fluidity or elasticity and drug entrapment. All of these lead to the different mechanisms of action with possible vesicle skin penetration reaching different depths, from surface assimilation to (rarely) the viable tissue and subsequent systemic absorption. So, we limit ourselves to explaining the possible mechanisms of liposomes promoting transdermal absorption of drugs. Firstly, fusion mechanism, the phospholipids contained in liposomes have a high compatibility with the lipids of the SC that allows lipophilic active molecules 'to flow' more easily [140]. At the surface of SC, the structure of liposomes break up, thereby enabling phospholipids to penetrate within the SC. This allows them to form a flat granular structure in a way that the drugs encapsulated in liposomes can easily enter the skin through the lipid granular space, while promoting skin permeation of the effective payload [141, 142]. Secondly, the hydration mechanism, wherein the liposomes can increase the moisture of the cuticle, promote hydration of the skin, change the orderlystructureof the lipid layer between the keratinocytes, reduce the density, and enhance the permeability of the drugs [143]. Thirdly, the penetrating mechanism, some researchers believe that the liposomes can penetrate through keratinocytes directly into the deep layer of skin. In addition, they can also enter the subcutaneous layer directly through the channels in the skin appendages to achieve transdermal effect [144].
3.2. TransfersomesThe transfersomes are formed by improving the component of liposomes. The surfactants embedded in the vesicle membrane facilitate a high degree of deformation so that they can penetrate the pores several times smaller than their own size [145]. The mechanism of transfersomes by which they efficiently penetrate the SC can be summed up in three aspects. Firstly, surfactants can penetrate into and interact with the SC and leads to swelling, after which the keratin turns into swollen uncoiled fibers, extracting lipids from the SC and disturbing its organization [146]. The crystalline and gel states of the long-chain molecules improve the fluidity of lipids, and produce or expand the hydrophilic voids, thus enhancing the permeability of the SC to hydrophilic substances [147]. Due to the "edge activators", the transfersomes can penetrate the so called "virtual channels" formed by intercellular lipid lamellae containing structural irregularities. Secondly, the water content in the SC (15%) is lower than that in the other layers of the epidermis (75%) resulting in a water gradient [41]. Under non-occlusive conditions, the skin hydration gradient is naturally formed during the transdermal process of transfersomes. The driving osmotic pressure of the gradient has nothing to do with the concentration, so it may be driven by the osmotic pressure difference. With the help of shape compression, it is possible to efficiently traverse a channel that is several times the size of its own [148, 149]. Due to the high deformability, transfersomes can squeeze through the gaps in SC. The theory of osmotic gradient and water combination has been accepted by the research community [150]. Thirdly, new work has proposed that the passage of transfersomes mainly occur via the hair follicle pathway [151].
3.3. EthosomesEthosomes are multilayer vesicles containing high concentrations of alcohol added to the conventional liposomes' prescription [30]. The mechanism of ethosomes enhancing the permeability is related to its structure and the high concentration of alcohol. The high concentration of alcohol increases the mobility of polar lipid heads of the lipid molecules, leading to increased lipid fluidity and flexibility of the membranes. It also decreases the density of the intercellular lipid domains, causing the ethosomes to deform in the course of transmission. They interact with the disturbed SC (altering the intercellular lipid lamella) as well as pass-through the deranged SC, and create their own paths to cross the SC and reach the deep skin layers to increase permeability [152]. Secondly, the high concentration of alcohol increases the solubility of the drug in the lipid layer, as it also dehydrates and defats the SC, which promotes permeation for a long time. At the same time, it can alter the tight arrangement of the lipid bilayers in the SC, reduce the phase-transition temperature, enhance the fusion of the ethosomes with the lipids of SC, and also promote the percutaneous absorption of the drugs. So, the higher the alcohol content is, the greater the permeation flux [153]. The release and absorption of the drug in the deep skin is the result of the fusion of ethosomes with the skin lipid, which is accompanied by the entire transdermal process [154].
3.4. NiosomesNiosomes can increase the residence time of drugs in both the SC and the epidermis, while reducing the systemic absorption of the drug. The mechanism of niosomal permeation is mainly realized by non-ionic surface-active agents. The transdermal properties of niosomes are similar to those of liposomes [155]. The close contact between the niosomes and the SC of the skin, and their adsorption and fusion on the surface of the skin leads to an increase in the thermodynamic activity gradient of the drug at the interface, leading to an increase in the permeation of lipophilic drugs [27, 156]. Secondly, niosomes improve the SC property by reducing the trans-epidermal water loss, which can increase SC hydration and loosen its closely-packed cellular structure and improve smoothness through replenishment of lost skin lipids [157]. Thirdly, the lipid bimolecular layer of niosomes act as a ratelimiting membrane and play a local slow-release effect. The amount of drug permeation is also related to the properties of the encapsulated drugs. When the drug is encapsulated by the niosomes, the circulation time of the drug in the body can be prolonged, thus enhancing the penetration of the drug to the target organ and increasing the curative effect [140, 158].
3.5. InvasomesInvasomes improve skin penetration of hydrophilic and lipophilic drugs. The transdermal mechanism of the invasomes are concluded as below. One part of the vesicles is fragmented during their penetration into the upper skin layers. The released terpenes and phospholipids have the potential to increase the penetration of many drugs by disrupting the tight lipid packing of the SC, which acts as penetration enhancers. The deformability of invasomes is also associated with ethanol [159]. Some small intact invasomes may penetrate the SC without being fragmented because of the transepidermal osmotic gradient and the disturbed organization of SC lipids [160]. They can also achieve this by following some small hydrophilic channels existing in the intercellular space of the SC. Invasomes may also follow the hair follicle transport pathway, as reported [161].
3.6. Lipid nanoparticles (SLN and NLC)The possible mechanisms of lipid nanoparticles promoting percutaneous permeation of drugs are as follows: Lipid nanoparticles have good skin adhesion, which promotes contact between drugs and SC, and forms a mono-layered lipid film [162]. Lipid nanoparticles fuse and deform by capillary force, and form a hydrophobic film on the surface of the skin, both of which results in an 'occlusion effect'. This effect reduces the natural evaporation of water on the skin surface, increases the skin hydration, with a reduction in keratinocyte packing, and enlargement of the inter-keratinocyte gap that enhances deeper skin penetration of drugs [163]. Secondly, in addition, the smaller the particle size, the better the skin penetration. The small particle size may provide a higher specific surface area, and can enhance the 'occlusive effect' to promote transdermal penetration [164]. Thirdly, the nonionic surfactants in the composition of lipid nanoparticles also play a role in promoting permeability, because they enhance thermodynamic stability of SLN and NLC, and cause skin structure disruption [165]. The mechanism may also be affected by parameters such as the carrier itself, the type and concentration of the lipid, and the drug localization in the nanoparticle structure [166]. Fourthly, as for SLN, they have the reservoir effect from the skin surface and SC, because they have remained on the skin surface and in the SC, which forms the drug reservoir in the upper layers of the skin. They release the active substance in a biphasic regimen- an initial burst release from the surface of the particles and an aqueous phase, followed by a reservoir effect [167]. According to some reports, the drug in the NLC remains in the liquid lipid surrounded by the solid lipid. This arrangement gives the drug some degree of mobility, offers stability even when the solid lipid undergoes polymorphic changes [168]. In short, the skin adhesion and 'occlusion effect' of the lipid nanoparticles are the main mechanisms promoting permeability.
3.7. Polymeric nanoparticlesThe mechanisms of drug permeation promoted by polymeric NP are as follows: Firstly, complete polymeric NP are too large to spread passively through the SC directly into the deep position of the skin. However, they can be assembled on the surface of the skin or hair follicle, then taken to a drug reservoir, for gradual release of drugs from the nanoparticles. This results in a higher local concentration of the drug contained in the polymeric NP, as the drug diffuses into the active layer of the skin at the concentration gradient [169, 170]. Secondly, the integrity of skin and the physicochemical properties of nanoparticles affect the permeability of polymeric NP. Therefore, if the integrity of the skin can be changed appropriately, it could facilitate the percutaneous penetration of the drugs in the nanoparticles [171]. Thirdly, the surface charge of polymeric NP affects their permeability. The highly cationic components increase the surface charge, so the interaction with the negatively-charged skin surface is also much stronger leading to increase in drug release from the nanoparticles [172]. Fourthly, the permeation of polymeric NP is further enhanced through the hair follicle to enter the skin [173].
3.8. NanoemulsionsNE promote drug transdermal penetration through the following mechanisms: Firstly, the composition of NE such as the surface active agent, cosurfactant and oil phase, may play a role in promoting permeability by disturbing the arrangement of the lipids in the SC. This results in increase in the amount of transdermal permeation of the drug [174]. Secondly, because of the unique structure of NE, both liposoluble and water-soluble drugs can be solubilized significantly (especially liposoluble drugs). This leads to an increase in the permeable concentration gradient inside and outside of the skin, which actually enhances the penetration of drugs [175]. Thirdly, the storage function of the inner phase maintains a constant driving force of drug diffusion from the external phase to the skin, and prolongs the drug absorption [176]. Fourthly, NE expands the connections between SC by using the powerful combination with water to produce a faster percutaneous passage through the keratinocytes. They also release the collagen fibers in the dermis to a certain extent [177]. Fifth, the NE has a small particle size, a large specific surface area, and a low surface tension, which enables them good wettability and increased contact with the skin, thereby benefitting the penetrability of the drug [178].
4. ConclusionIn this review, we systematically summarized the components, characteristics, and transdermal mechanisms, as well as the application of liposomes, transfersomes, ethosomes, niosomes, invasomes, solid lipid nanoparticles, nanostructured lipid carriers, polymeric nanoparticles and nano-emulsions for TDDS. This review offers a complete picture of TDDS and aims to improve the understanding of the nano-formulations used in TDDS. Furthermore, it lays a foundation for future research and promotes the clinical application of TDDS.
AcknowledgmentsThis work was supported by the Postdoctoral Innovation Talents Support Program (No. BX20180207) and the National Nature Science Foundation of China (No. 81502722).
| [1] |
H. Trommer, R.H. Neubert, Skin Pharmacol. Physio. 19 (2006) 106-121. DOI:10.1159/000091978 |
| [2] |
Z.A.M. Yasin, F. Lbrahim, N.N. Rashid, et al., Curr. Pharm. Biotechnol. 18 (2017) 864-876. |
| [3] |
L. Ventrelli, L.M. Strambini, G. Barillaro, Adv. Healthcare Mater. 4 (2015) 2606-2640. DOI:10.1002/adhm.201500450 |
| [4] |
C.Y. Zhao, P. Quan, C. Liu, et al., Acta Pharm. Sin. B 6 (2016) 623-628. DOI:10.1016/j.apsb.2016.05.012 |
| [5] |
C. Kodiweera, Y. Yang, A.L. Bunge, J. Pharm. Sci. 17 (2017) 1131-1142. |
| [6] |
M. Ma, H.J. Di, H. Zhang, J.H. Yao, et al., Drug Dev. Ind. Pharm. 43 (2017) 2055-2063. DOI:10.1080/03639045.2017.1371730 |
| [7] |
G. Cevc, U. Vierl, J. Control. Release 141 (2010) 277-299. DOI:10.1016/j.jconrel.2009.10.016 |
| [8] |
M. Armengot-Carbo, A. Hernandez-Martin, A. Torrelo, Actas Dermosifiliogr. 106 (2015) 86-95. DOI:10.1016/j.ad.2013.10.019 |
| [9] |
B. Illel, H. Schaefer, J. Wepierre, et al., J. Pharm. Sci. 80 (1991) 424-427. DOI:10.1002/jps.2600800505 |
| [10] |
F. Erdo, N. Hashimoto, G. Karvaly, et al., J. Control.Release 233 (2016) 147-161. DOI:10.1016/j.jconrel.2016.05.035 |
| [11] |
Y. Hao, W. Li, X.L. Zhou, et al., J. Biomed. Nanotechnol. 13 (2017) 1581-1597. DOI:10.1166/jbn.2017.2474 |
| [12] |
B. Barry, J. Control. Release 6 (1987) 85-88. DOI:10.1016/0168-3659(87)90066-6 |
| [13] |
H. Marwah, T. Garg, A.K. Goyal, et al., Drug Deliv. 23 (2016) 564-578. DOI:10.3109/10717544.2014.935532 |
| [14] |
S. Meng, C. Zhang, W. Shi, et al., Eur. J. Pharm. Sci. 92 (2016) 49-54. DOI:10.1016/j.ejps.2016.04.033 |
| [15] |
K. Singh, N. Arora, T. Garg, Am. J. Pharm. Tech. Res. 2 (2012) 113-126. |
| [16] |
O. Singh, T. Garg, G. Rath, et al., J. Pharm. (Cairo) 2014 (2014) 1-47. |
| [17] |
G. Cevc, Exp. Opin. Invest Drugs 6 (1997) 1887-1901. DOI:10.1517/13543784.6.12.1887 |
| [18] |
Y.H. Weng, J. Liu, S.B. Jin, et al., Acta Pharm. Sin. B 7 (2017) 281-291. DOI:10.1016/j.apsb.2016.09.001 |
| [19] |
J. Liu, Q.W. Chen, K. Wu, Chin. Chem. Lett. 28 (2017) 1631-1639. DOI:10.1016/j.cclet.2017.04.022 |
| [20] |
K. Aziz, N. Ahmad, K. Kohli, J. Nanosci. Nanotechnol. 17 (2017) 4573-4583. DOI:10.1166/jnn.2017.14108 |
| [21] |
P.L. Honeywell-Nguyen, J.A. Bouwstra, Drug Discov. Today Technol. 2 (2005) 67-74. DOI:10.1016/j.ddtec.2005.05.003 |
| [22] |
G. Gregoriadis, A.T. Florence, Drugs 45 (1993) 15-48. |
| [23] |
Z. Li, H.Y. Li, C.F. Wang, et al., Acta Pharm. Sin. B 6 (2016) 344-351. DOI:10.1016/j.apsb.2016.03.003 |
| [24] |
A.D. Bangham, Bioessays. 17 (1995) 1081-1088. |
| [25] |
M. Mezei, V. Gulasekharam, Life Sci. 26 (1980) 1473-1477. |
| [26] |
C. Sinico, A.M. Fadda, Exp. Opin. Drug Deliv. 6 (2009) 813-825. |
| [27] |
L. Tavano, P. Alfano, R. Muzzalupo, et al., Colloids Surf. B-Biointerfaces 87 (2011) 333-339. DOI:10.1016/j.colsurfb.2011.05.041 |
| [28] |
G. Cevc, G. Blume, Biochim. Biophys. Acta 1104 (1992) 226-232. DOI:10.1016/0005-2736(92)90154-E |
| [29] |
B.A. van den Bergh, J. Vroom, H. Gerritsen, et al., Biochim. Biophys. Acta 1461 (1999) 155-173. DOI:10.1016/S0005-2736(99)00176-5 |
| [30] |
E. Touitou, N. Dayan, L. Bergelson, et al., J. Control. Release 3 (2000) 403-418. |
| [31] |
N. Dragicevic-Curic, D. Scheglmann, V. Albrecht, et al., J. Control. Release 127 (2008) 59-69. DOI:10.1016/j.jconrel.2007.12.013 |
| [32] |
M. Mezei, V. Gulasekharam, Life Sci. 26 (1980) 1473-1477. |
| [33] |
J.O. Eloy, M. Claro de Souza, R. Petrilli, et al., Colloids Surf. B-Biointerfaces 123 (2014) 345-363. DOI:10.1016/j.colsurfb.2014.09.029 |
| [34] |
Ž. Vanić, A. Hafner, M. Bego, et al., Drug Dev. Ind. Pharm. 39 (2013) 481-488. DOI:10.3109/03639045.2012.670247 |
| [35] |
M. Ning, Z. Gu, H. Pan, et al., Indian J. Exp. Biol. 43 (2005) 150-157. |
| [36] |
J.S. Seong, M.E. Yun, S.N. Park, Carbohydr. Polym. 181 (2018) 659-667. DOI:10.1016/j.carbpol.2017.11.098 |
| [37] |
G. Oliveira, J.C. Leverett, M. Emamzadeh, et al., Int. J. Pharm. 464 (2014) 145-151. DOI:10.1016/j.ijpharm.2014.01.012 |
| [38] |
M.L. Manca, P. Matricardi, C. Cencetti, et al., Int. J. Pharm. 505 (2016) 204-211. DOI:10.1016/j.ijpharm.2016.04.008 |
| [39] | |
| [40] |
M. Ashtikar, K. Nagarsekar, A. Fahr, J. Control. Release 242 (2016) 126-140. DOI:10.1016/j.jconrel.2016.09.008 |
| [41] |
E.L. Romero, M.J. Morilla, Int. J. Nanomed. Nanosurg. 8 (2013) 3171-3186. |
| [42] |
A. Kumar, K. Pathak, V. Bali, Drug Discov. Today 17 (2012) 1233-1241. DOI:10.1016/j.drudis.2012.06.013 |
| [43] |
N. Aggarwal, S. Goindi, Int. J. Pharm. 437 (2012) 277-287. DOI:10.1016/j.ijpharm.2012.08.015 |
| [44] |
W.S. Zheng, X.Q. Fang, L.L. Wang, et al., Int. J. Pharm. 436 (2012) 291-298. DOI:10.1016/j.ijpharm.2012.07.003 |
| [45] |
S. Duangjit, P. Opanasopit, T. Rojanarata, et al., AAPS Pharm. Sci. Tech. 14 (2013) 133-140. DOI:10.1208/s12249-012-9904-2 |
| [46] |
J.H. Choi, S.H. Cho, J.J. Yun, et al., J. Nanosci. Nanotechnol. 15 (2015) 5660-5662. DOI:10.1166/jnn.2015.10462 |
| [47] |
K. Milger, J.F. Felix, R. Voswinckel, et al., Pulm. Circ. 5 (2015) 305-312. DOI:10.1086/680355 |
| [48] |
K.M. Hosny, B.M. Aljaeid, Expert Opin. Drug Deliv. 11 (2014) 1015-1022. DOI:10.1517/17425247.2014.912212 |
| [49] |
S.M. Badr-Eldin, O.A. Ahmed, Drug Des. Dev. Ther. 10 (2016) 1323-1333. |
| [50] |
R. Rajan, D.T. Vasudevan, J. Adv. Pharm. Technol. Res. 3 (2012) 112-116. DOI:10.4103/2231-4040.97286 |
| [51] |
I. Scognamiglio, D. De Stefano, V. Campani, et al., Int. J. Pharm. 440 (2013) 179-187. DOI:10.1016/j.ijpharm.2012.08.009 |
| [52] |
A. Ahad, A.A. Al-Saleh, A.M. Al-Mohizea, et al., Biomed. Pharmacother. 89 (2017) 177-184. DOI:10.1016/j.biopha.2017.01.164 |
| [53] |
Y. Wo, Z. Zhang, Y.X. Zhang, et al., J. Nanosci. Nanotechnol. 11 (2011) 7840-7847. DOI:10.1166/jnn.2011.4741 |
| [54] |
C. Mbah, P.F. Builders, A.A. Attama, Expert. Opin. Drug Deliv. 11 (2014) 45-59. DOI:10.1517/17425247.2013.860130 |
| [55] |
Y.T. Zhang, L.N. Shen, Z.H. Wu, et al., Int. J. Pharm. 471 (2014) 449-452. DOI:10.1016/j.ijpharm.2014.06.001 |
| [56] |
B. Godin, E. Touitou, Curr. Drug Deliv. 2 (2005) 269-275. DOI:10.2174/1567201054367931 |
| [57] |
X. Zhu, F. Li, X. Peng, et al., Anesth. Analg. 117 (2013) 352-357. DOI:10.1213/ANE.0b013e3182937b74 |
| [58] |
C.C. Mbah, P.F. Builders, A.A. Attama, Expert Opin. Drug Deliv. 11 (2014) 45-59. |
| [59] |
S.A. Iizhar, I.A. Syed, R. Satar, et al., J. Adv. Res. 7 (2016) 453-461. DOI:10.1016/j.jare.2016.03.003 |
| [60] |
E.R. Bendas, M.I. Tadros, AAPS Pharm. Sci. Tech. 8 (2007) E1-E8. |
| [61] |
S. Shelke, S. Shahi, S. Jalalpure, et al., J. Liposome Res. 26 (2016) 313-323. DOI:10.3109/08982104.2015.1132232 |
| [62] |
F. Yao, M. Zhang, L. Chen, Acta Pharm. Sin. B 6 (2016) 20-25. DOI:10.1016/j.apsb.2015.07.009 |
| [63] |
L. Ge, X.R. You, Y. Zhang, et al., J. Biomed. Nanotechnol. 13 (2017) 931-945. DOI:10.1166/jbn.2017.2385 |
| [64] |
K. Li, S. Gao, B. Tian, et al., Curr. Drug Deliv. 15 (2018) 424-435. DOI:10.2174/1567201815666171207163010 |
| [65] |
S. Moghassemi, A. Hadjizadeh, J. Control. Release 185 (2014) 22-36. DOI:10.1016/j.jconrel.2014.04.015 |
| [66] |
I.P. Kaur, A. Garg, A.K. Singla, et al., Int. J. Pharm. 269 (2004) 1-14. |
| [67] |
M.Z. Ahmad, A.A. Mohammed, M.M. Ibrahim, Pharm. Dev. Technol. 22 (2017) 302-311. DOI:10.3109/10837450.2015.1135344 |
| [68] |
S. Meng, Z. Chen, L. Yang, et al., Int. J. Nanomedicine 8 (2013) 3051-3060. |
| [69] |
A. Manosroi, C. Chankhampan, W. Manosroi, et al., Eur. J. Pharm. Sci. 48 (2013) 474-483. DOI:10.1016/j.ejps.2012.12.010 |
| [70] |
P. Arunothayanun, I.F. Uchegbu, A.T. Florence, J. Pharm. Pharmacol. 51 (1999) 651-657. |
| [71] |
H. Abdelkader, Z. Wu, R. Al-Kassas, et al., Int. J. Pharm. 433 (2012) 142-148. DOI:10.1016/j.ijpharm.2012.05.011 |
| [72] |
S. Ghosh, B. Mukherjee, S. Chaudhuri, et al., AAPS Pharm. Sci. Tech. 19 (2018) 1320-1336. DOI:10.1208/s12249-017-0939-2 |
| [73] |
C. Marianecci, L. Di Marzio, F. Rinaldi, et al., Adv. Colloid Interface Sci. 205 (2013) 187-206. |
| [74] |
Y.S. Tu, D.M. Sun, J.J. Zhang, et al., J. Microencapsulation 31 (2014) 307-316. DOI:10.3109/02652048.2013.843727 |
| [75] |
D.G. Smith, Am. J. Manag. Care 13 (2007) S68-S71. |
| [76] |
L. Amati, M. Chiloiro, E. Jirillo, et al., Curr. Pharm. Des. 13 (2007) 3696-3700. DOI:10.2174/138161207783018563 |
| [77] |
I.C. Gelissen, H.L. Nguyen, D.K. Tiao, et al., Paediatr. Drugs 16 (2014) 417-423. DOI:10.1007/s40272-014-0087-z |
| [78] |
A.S. Zidan, K.M. Hosny, O.A. Ahmed, et al., Drug Deliv. 23 (2016) 1536-1549. |
| [79] |
S.K. Kumar, M. Rudrapal, B. Mazumder, et al., Drug Deliv. 22 (2015) 1043-1052. DOI:10.3109/10717544.2013.861041 |
| [80] |
J. Donnerer, R. Amann, R. Schuligoi, et al., Arch. Pharmacol. 342 (1990) 357-361. |
| [81] |
N. Dragicevic-Curic, D. Scheglmann, V. Albrecht, et al., Colloids Surf. B-Biointerfaces 70 (2009) 198-206. DOI:10.1016/j.colsurfb.2008.12.030 |
| [82] |
S.M. Shah, M. Ashtikar, A.S. Jain, et al., Int. J. Pharm. 490 (2015) 391-403. DOI:10.1016/j.ijpharm.2015.05.042 |
| [83] |
M. Dwivedi, V. Sharma, K. Pathak, Drug Dev. Ind. Pharm. 43 (2017) 293-304. DOI:10.1080/03639045.2016.1239628 |
| [84] |
G.R. Qadri, A. Ahad, M. Aqil, et al., Artif. Cells Nanomed. Biotechnol. 45 (2016) 139-145. |
| [85] |
M. Kamran, A. Ahad, M. Aqil, et al., Int. J. Pharm. 505 (2016) 147-158. DOI:10.1016/j.ijpharm.2016.03.030 |
| [86] |
S.Z. Chen, X.H. Hao, X.J. Liang, et al., J. Biomed. Nanotechnol. 12 (2016) 1-27. DOI:10.1166/jbn.2016.2122 |
| [87] |
L. Yang, J. He, Y. Wen, et al., J. Biomed. Nanotechnol. 12 (2016) 1348-1373. DOI:10.1166/jbn.2016.2284 |
| [88] |
A. Ranganathan, J. Campo, J. Myerson, et al., J. Biomed. Nanotechnol. 13 (2017) 737-745. DOI:10.1166/jbn.2017.2392 |
| [89] |
K. Zhang, P.P. Yang, J.P. Zhang, et al., Chin. Chem. Lett. 28 (2017) 1808-1816. DOI:10.1016/j.cclet.2017.07.001 |
| [90] |
G.M. Soliman, Int. J. Pharm. 523 (2017) 15-32. DOI:10.1016/j.ijpharm.2017.03.019 |
| [91] |
K.B. Wang, C.M. Yang, J.J. Ye, et al., J. Biomed. Nanotechnol. 13 (2017) 717-726. DOI:10.1166/jbn.2017.2378 |
| [92] |
J. Pardeike, A. Hommoss, R.H. Müller, Int. J. Pharm. 366 (2009) 170-184. DOI:10.1016/j.ijpharm.2008.10.003 |
| [93] |
M.C. Teixeira, C. Carbone, E.B. Souto, Prog. Lipid Res. 68 (2017) 1-11. DOI:10.1016/j.plipres.2017.07.001 |
| [94] |
P. Siafaka, M. Betsiou, A. Tsolou, et al., J. Mater. Sci. Mater. Med. 26 (2015) 275-287. DOI:10.1007/s10856-015-5609-x |
| [95] |
S.A. Wissing, O. Kayser, R.H. Müller, Adv. Drug Deliv. Rev. 56 (2004) 1257-1272. DOI:10.1016/j.addr.2003.12.002 |
| [96] |
E. Winter, C.D. Pizzol, C. Locatelli, et al., J. Nanosci. Nanotechnol. 16 (2016) 1321-1330. DOI:10.1166/jnn.2016.11667 |
| [97] |
J.H. Sun, S.Z. Zhang, S.J. Jiang, et al., J. Biomed. Nanotechnol. 12 (2016) 1709-1723. DOI:10.1166/jbn.2016.2285 |
| [98] |
Y.T. Zhang, M.Q. Han, L.N. Shen, et al., J. Biomed. Nanotechnol. 11 (2015) 351-361. DOI:10.1166/jbn.2015.1902 |
| [99] |
C.E.M. Griffiths, J.M. van der Walt, D.M. Ashcroft, et al., Br. J. Dermatol. 177 (2017) e4-e7. DOI:10.1111/bjd.15610 |
| [100] |
L.C. Coates, P.S. Helliwell, J. Rheumatol. 43 (2016) 356-361. DOI:10.3899/jrheum.150614 |
| [101] |
S. Dogra, R. Mahajan, Clin. Exp. Dermatol. 38 (2013) 573-588. DOI:10.1111/ced.2013.38.issue-6 |
| [102] |
S.R. Feldman, Y. Zhao, P. Navaratnam, et al., J. Manag. Care Spec. Pharm. 21 (2015) 201-209. |
| [103] |
M. Ferreira, L. Barreiros, M.A. Segundo, et al., Colloids Surf. B-Biointerfaces 159 (2017) 23-29. DOI:10.1016/j.colsurfb.2017.07.080 |
| [104] |
R.H. Müller, R.D. Petersen, A. Hommoss, et al., Adv. Drug Deliv. Rev. 59 (2007) 522-530. DOI:10.1016/j.addr.2007.04.012 |
| [105] |
S. Doktorovova, E.B. Souto, Expert Opin. Drug Deliv. 6 (2009) 165-176. DOI:10.1517/17425240802712590 |
| [106] |
L. Aliasgharlou, S. Ghanbarzadeh, H. Azimi, et al., Adv. Pharm. Bull. 6 (2016) 581-587. DOI:10.15171/apb.2016.072 |
| [107] |
C. Vitorino, A. Almeida, J. Sousa, et al., Eur. J. Pharm. Biopharm. 86 (2014) 133-144. DOI:10.1016/j.ejpb.2013.12.004 |
| [108] |
U. Leiter, C. Garbe, Adv. Exp. Med. Biol. 624 (2008) 89-103. DOI:10.1007/978-0-387-77574-6 |
| [109] |
F.F. Zhong, X.L. Yuan, J.Z. Zhao, et al., Sci. China Chem. 59 (2016) 70-77. |
| [110] |
H.D. Cui, D.H. Hu, J.N. Zhang, et al., Chin. Chem. Lett. 28 (2017) 1391-1398. DOI:10.1016/j.cclet.2016.12.038 |
| [111] |
A. Qidwai, S. Khan, S. Md, et al., Drug Deliv. 23 (2016) 1476-1485. DOI:10.3109/10717544.2016.1165310 |
| [112] |
S. Babaei, S. Ghanbarzadeh, Z.M. Adib, et al., Pharmazie 71 (2016) 247-251. |
| [113] |
H. Strongman, S. Christopher, M. Majak, et al., BMJ Open Diabetes Res. Care 6 (2018) 1-12. |
| [114] |
A. Esteghamati, R. Aziz, M. Ebadi, et al., Exp. Clin. Endocrinol. Diabetes 123 (2015) 289-295. DOI:10.1055/s-00000017 |
| [115] |
S. Alam, M. Aslam, A. Khan, et al., Drug Deliv. 23 (2016) 601-609. DOI:10.3109/10717544.2014.923958 |
| [116] |
S. Vrignaud, J.P. Benoit, P. Saulnier, Biomaterials 32 (2011) 8593-8604. DOI:10.1016/j.biomaterials.2011.07.057 |
| [117] |
K.N. Zhu, Z.Y. Zhu, H.O. Zhou, et al., Chin. Chem. Lett. 28 (2017) 1276-1284. DOI:10.1016/j.cclet.2017.03.020 |
| [118] |
C.Y. Hu, Z. Chen, S.J. Wu, et al., Chin. Chem. Lett. 28 (2017) 1905-1909. DOI:10.1016/j.cclet.2017.07.020 |
| [119] |
S.H. Voon, S.X. Tiew, C.S. Kue, et al., J. Biomed. Nanotechnol. 12 (2016) 1431-1452. DOI:10.1166/jbn.2016.2263 |
| [120] |
H. Zhao, Y.L. Wang, J.R. Peng, et al., J. Biomed. Nanotechnol. 13 (2017) 427-436. DOI:10.1166/jbn.2017.2357 |
| [121] |
C. Pegoraro, S. MacNeil, G. Battaglia, Nanoscale 4 (2012) 1881-1894. DOI:10.1039/c2nr11606e |
| [122] |
J.M. Lee, K.J. Lee, M. Kang, et al., J. Nanosci. Nanotechnol. 17 (2017) 7515-7519. DOI:10.1166/jnn.2017.14798 |
| [123] |
I. Savoye, C.M. Olsen, D.C. Whiteman, et al., J. Epidemiol. 58 (2018) 27-33. |
| [124] |
R. Edlich, K.L. Winters, H.W. Lim, et al., J. Long. Eff. Med. Implants 14 (2004) 317-340. DOI:10.1615/JLongTermEffMedImplants.v14.i4 |
| [125] |
P.K. Shetty, V. Venuvanka, H.V. Jagani, et al., Int. J. Nanomed. 10 (2015) 6477-6491. |
| [126] |
P. Batheja, L. Sheihet, J.J. Kohn, et al., J. Control. Release 149 (2011) 159-167. DOI:10.1016/j.jconrel.2010.10.005 |
| [127] |
T. Zhang, D.Q. Xu, J.M. Chen, et al., Chin. Chem. Lett. 27 (2016) 441-446. DOI:10.1016/j.cclet.2015.12.028 |
| [128] |
J. Wang, G.S. Williamson, M.G. Lancina, et al., J. Biomed. Nanotechnol. 13 (2017) 1089-1096. DOI:10.1166/jbn.2017.2436 |
| [129] |
Y. Singh, J.G. Meher, K. Raval, et al., J. Control. Release 252 (2017) 28-49. DOI:10.1016/j.jconrel.2017.03.008 |
| [130] |
K. Bouchemal, S. Brainçon, E. Perrier, et al., Int. J. Pharm. 280 (2004) 241-251. DOI:10.1016/j.ijpharm.2004.05.016 |
| [131] |
N. Anton, J.P. Benoit, P. Saulnier, J. Control. Release 128 (2008) 185-199. DOI:10.1016/j.jconrel.2008.02.007 |
| [132] |
Y. Chang, L. McLandsborough, D.J. McClements, J. Agric. Food Chem. 60 (2013) 12056-12063. |
| [133] |
L.L. Kong, E. Amstad, M.T. Hai, et al., Chin. Chem. Lett. 28 (2017) 1897-1900. DOI:10.1016/j.cclet.2017.07.017 |
| [134] |
P. Sinha, S. Srivastava, N. Mishra, et al., Drug Dev. Ind. Pharm. 42 (2016) 1434-1435. DOI:10.3109/03639045.2016.1141931 |
| [135] |
B. Kwasigroch, E. Escribano, M.D.C. Morán, et al., Int. J. Pharm. 515 (2016) 749-756. DOI:10.1016/j.ijpharm.2016.11.016 |
| [136] |
A. Kaur, S.S. Katiyar, V. Kushwah, et al., Nanomedicine 13 (2017) 1473-1482. DOI:10.1016/j.nano.2017.02.009 |
| [137] |
F. Shakeel, W. Ramadan, Colloids Surf. B-Biointerfaces 75 (2010) 356-362. DOI:10.1016/j.colsurfb.2009.09.010 |
| [138] |
C.B. de Mattos, D.F. Argenta, G. de Lima Melchiades, et al., Int. J. Nanomedicine 10 (2015) 5229-5542. |
| [139] |
C. Artman, J. Roding, M. Ghyczy, et al., Arzneimittelforschung 40 (1990) 1365-1368. |
| [140] |
A. Laouini, C. Charcosset, H. Fessi, et al., Colloids Surf. B-Biointerfaces 112 (2013) 272-278. DOI:10.1016/j.colsurfb.2013.07.066 |
| [141] |
M. Kirjavainen, A. Urtti, I. Jaaskelainen, et al., Biochim. Biophys. Acta 1304 (1996) 179-189. DOI:10.1016/S0005-2760(96)00126-9 |
| [142] |
A. Kato, Y. Ishibashi, Y. Miyake, J. Pharm. Pharmacol. 39 (1987) 399-400. DOI:10.1111/jphp.1987.39.issue-5 |
| [143] |
M. Kirjavainen, J. Monkkonen, M. Saukkosaari, et al., J. Control. Release 58 (1999) 207-214. DOI:10.1016/S0168-3659(98)00152-7 |
| [144] |
G.M. El Maghraby, A.C. Williams, B.W. Barry, J. Pharm. Pharmacol. 58 (2006) 415-429. DOI:10.1211/jpp.58.4.0001 |
| [145] |
M.E. Planas, P. Gonzalez, L. Rodriguez, et al., Anesth. Analg. 75 (1992) 615-621. |
| [146] |
D. Singh, M. Pradhan, M. Nag, et al., Artif. Cells Nanomed. Biotechnol. 43 (2015) 282-290. DOI:10.3109/21691401.2014.883401 |
| [147] |
S. Duangjit, B. Pamornpathomkul, P. Opanasopit, et al., Int. J. Nanomed. 9 (2014) 2005-2017. |
| [148] |
K. Priyanka, S.A. Singh, Curr. Drug Targets 15 (2014) 184-198. |
| [149] |
M.L. González-Rodríguez, C.M. Arroyo, M.J. Cózar-Bernal, et al., Drug Dev. Ind. Pharm. 42 (2016) 1683-1694. DOI:10.3109/03639045.2016.1165691 |
| [150] |
M.J. Choi, H.I. Maibach, Int. J. Cosmet. Sci. 27 (2005) 211-221. DOI:10.1111/ics.2005.27.issue-4 |
| [151] |
T. Subongkot, N. Wonglertnirant, P. Songprakhon, et al., Int. J. Pharm. 441 (2013) 151-161. DOI:10.1016/j.ijpharm.2012.12.003 |
| [152] |
K. Priyanka, S. Singh, Curr. Drug Targets 15 (2014) 184-198. DOI:10.2174/1389450115666140113100338 |
| [153] |
G. Li, Y. Fan, C. Fan, et al., Eur. J. Pharm. Biopharm. 82 (2012) 49-57. DOI:10.1016/j.ejpb.2012.05.011 |
| [154] |
M.M. Elsayed, O.Y. Abdallah, V.F. Naggar, et al., Int. J. Pharm. 332 (2007) 1-16. |
| [155] |
J.A. Bouwstra, P.L. Honeywell-Nguyen, G.S. Gooris, et al., Prog. Lipid Res. 42 (2003) 1-36. DOI:10.1016/S0163-7827(02)00028-0 |
| [156] |
N. Mali, S. Darandale, P. Vavia, Drug Deliv. Transl. Res. 3 (2013) 587-592. DOI:10.1007/s13346-012-0083-1 |
| [157] |
A. Ossmann, S. Kranz, G. Andre, et al., Clin. Oral Investig. 19 (2015) 373-384. DOI:10.1007/s00784-014-1271-9 |
| [158] |
G. loele, L. Tavano, M. De Luca, et al., Int. J. Pharm. 494 (2015) 490-497. DOI:10.1016/j.ijpharm.2015.08.053 |
| [159] |
H.K. Vaddi, P.C. Ho, Y.W. Chan, et al., J. Control. Release 81 (2002) 121-133. DOI:10.1016/S0168-3659(02)00057-3 |
| [160] |
C. Sinico, A.M. Fadda, Expert Opin. Drug Deliv. 6 (2009) 813-825. DOI:10.1517/17425240903071029 |
| [161] |
D.D. Verma, S. Verma, K.J. McElwee, et al., Eur. J. Dermatol. 14 (2004) 332-338. |
| [162] |
R.H. Müller, M. Radtke, S.A. Wissing, Adv. Drug Deliv. Rev. 54 (2002) S131-S155. DOI:10.1016/S0169-409X(02)00118-7 |
| [163] |
S.A. Wissing, A. Lippacher, R.H. Müller, J. Cosmet. Sci. 52 (2001) 313-324. |
| [164] |
C. Vitorino, J. Almeida, L.M. Gonçalves, et al., J. Control. Release 167 (2013) 301-314. DOI:10.1016/j.jconrel.2013.02.011 |
| [165] |
Q. Xia, A. Saupe, R.H. Müller, et al., Int. J. Cosmet. Sci. 29 (2007) 473-482. DOI:10.1111/ics.2007.29.issue-6 |
| [166] |
Z. Mardhiah Adib, S. Ghanbarzadeh, M. Kouhsoltani, et al., Adv. Pharm. Bull. 6 (2016) 31-36. DOI:10.15171/apb.2016.06 |
| [167] |
L.B. Jensen, K. Petersson, H.M. Nielsen, Eur. J. Pharm. Biopharm. 79 (2011) 68-75. DOI:10.1016/j.ejpb.2011.05.012 |
| [168] |
A. Beloqui, M.A. Solinis, A. Rodriguez-Gascon, et al., Nanomedicine 12 (2016) 143-161. DOI:10.1016/j.nano.2015.09.004 |
| [169] |
F. Rancan, D. Papakostas, S. Hadam, et al., Pharm. Res. 26 (2009) 2027-2036. DOI:10.1007/s11095-009-9919-x |
| [170] |
A.C. Watkinson, A.L. Bunge, J. Hadgraft, et al., Pharm. Res. 30 (2013) 1943-1946. DOI:10.1007/s11095-013-1073-9 |
| [171] |
E. Kimura, Y. Kawano, H. Todo, et al., Biol. Pharm. Bull. 35 (2012) 1476-1486. DOI:10.1248/bpb.b12-00103 |
| [172] |
K.I. McConnell, S. Shamsudeen, I.M. Meraz, et al., J. Biomed. Nanotechnol. 12 (2016) 154-164. DOI:10.1166/jbn.2016.2134 |
| [173] |
P.R. Desai, S. Marepally, A.R. Patel, et al., J. Control. Release 170 (2013) 51-63. DOI:10.1016/j.jconrel.2013.04.021 |
| [174] |
F. Shakeel, S. Baboota, A. Ahuja, et al., Pharmazie 64 (2009) 258-259. |
| [175] |
J. Lawrencem, D. Reesg, Adv. Drug Deliv. Rev. 45 (2000) 89-121. DOI:10.1016/S0169-409X(00)00103-4 |
| [176] |
S. Khurana, P.N.K. Jain, M.S. Bedi, Life Sci. 92 (2013) 383-392. DOI:10.1016/j.lfs.2013.01.005 |
| [177] |
P. Santos, A.C. Watkinson, J. Hadgraft, et al., Skin Pharmacol. Physiol. 21 (2008) 246-259. DOI:10.1159/000140228 |
| [178] |
H. Zhou, Y. Yue, G. Liu, et al., Nanoscale Res. Lett. 5 (2009) 224-230. |
| [179] |
C. Sinico, M. Manconi, M. Peppi, et al., J. Control. Release 103 (2005) 123-136. DOI:10.1016/j.jconrel.2004.11.020 |
| [180] |
H.C. Korting, H. Zienicke, M. Schäfer-Korting, et al., J. Clin. Pharmacol. 39 (1990) 349-351. DOI:10.1007/BF00315408 |
| [181] |
G. Cevc, G. Blume, Biochim. Biophys. Acta 1514 (2001) 191-205. DOI:10.1016/S0005-2736(01)00369-8 |
| [182] |
G. Cevc, G. Blume, Biochim. Biophys. Acta 1614 (2003) 156-164. DOI:10.1016/S0005-2736(03)00172-X |
| [183] |
V. Dubey, D. Mishra, M. Nahar, et al., Nanomedicine 6 (2010) 590-596. DOI:10.1016/j.nano.2010.01.002 |
| [184] |
P. Verma, K. Pathak, Nanomedicine 8 (2012) 489-496. DOI:10.1016/j.nano.2011.07.004 |
| [185] |
A. Dwivedi, A. Mazumder, L.T. Fox, et al., Int. J. Pharm. 503 (2016) 1-7. DOI:10.1016/j.ijpharm.2016.02.041 |
| [186] |
K.K. Patel, P. Kumar, H.P. Thakkar, AAPS Pharm. Sci. Tech. 13 (2012) 1502-1510. DOI:10.1208/s12249-012-9871-7 |
| [187] |
G.R. Qadri, A. Ahad, M. Aqil, et al., Artif. Cells Nanomed. Biotechnol. 45 (2017) 139-145. DOI:10.3109/21691401.2016.1138486 |
| [188] |
N. Dragicevic-Curic, S. Gräfe, V. Albrecht, et al., J. Photochem. Photobiol. B 91 (2008) 41-50. DOI:10.1016/j.jphotobiol.2008.01.009 |
| [189] |
Y. Gu, M. Yang, X. Tang, et al., J. Nanobiotechnol. 16 (2018) 68-82. DOI:10.1186/s12951-018-0389-3 |
| [190] |
S. El-Housiny, M.A. Shams Eldeen, Y.A. El-Attar, et al., Drug Deliv. 25 (2018) 78-90. DOI:10.1080/10717544.2017.1413444 |
| [191] |
E. Lason, E. Sikora, M. Miastkowska, et al., Acta Biochim. Pol. 65 (2018) 437-442. |
| [192] |
Y. Yue, D. Zhao, Q. Yin, Biomed. Pharmacother. 98 (2018) 813-820. DOI:10.1016/j.biopha.2017.12.103 |
| [193] |
I. Takeuchi, Y. Hida, K. Makino, Biomed. Mater. Eng. 29 (2018) 217-228. |
| [194] |
K. Yu, Y. Wang, T. Wan, et al., Int. J. Nanomed. 13 (2017) 129-142. |
| [195] |
P. Bakshi, Y. Jiang, T. Nakata, et al., J. Pharm. Sci. 11 (2018) 2883-2890. |
| [196] |
T. Zanela da Silva Marques, R. Santos-Oliveira, L. Betzler de Oliveira de Siqueira, et al., Int. J. Nanomed. 13 (2018) 2827-2837. |