 2018, Vol. 29
2018, Vol. 29 


 ,
Haojie Wanga,b,
Ruoyu Fana,b,
Haimin Zhanga,
Guozhong Wanga,
Huijun Zhaoa,c,*
,
Haojie Wanga,b,
Ruoyu Fana,b,
Haimin Zhanga,
Guozhong Wanga,
Huijun Zhaoa,c,*
b University of Science and Technology of China, Hefei 230026, China;
c Centre for Clean Environment and Energy, Gold Coast Campus, Griffith University, Queensland 4222, Australia
Recently, the production of fuels [1] and chemicals [2-4] from the biomass has attracted much attention due to the limited fossil fuel resources and global warming issues. Furfural (FAL), obtained by acid-catalyzed dehydration of xylose, has been viewed as one of the key platform molecules in biomass conversion [5]. The production of FAL has already been commercialized with the production of 105 tons/year, offering a rich and promising building block and/or platform for synthesis of important non-petroleumderived fuels and chemicals, such as furfuryl alcohol (FOL), tetrahydrofurfuryl alcohol (THFOL), 2-methylfuran (2-MF) through hydrogenation or hydrodeoxygenation [6]. The as-obtained alcohols, like FOL and THFOL, have been widely used as green solvents or important intermediates in the manufacture of chemical products, such as resins, lubricants, adhesives, fibers and wetting agents [7]. To obtain the THFOL with high yield, a twostep catalytic hydrogenation of FAL via the intermediate of FOL has usually been adopted over the hybrid catalysts composed of Cu-Cr and noble metal. Therefore, researchers aim at developing efficient catalyst to highly selective production of THFOL from FAL hydrogenation in one-step eco-friend and low-cost system [8]. Recently, some catalysts have been used to produce THFOL from FAL with a satisfactory result [9, 10], but the use of the precious metal and harsh reaction conditions may become some important factors to hinder their industrialization of FAL hydrogenation.
Nickel-based catalysts has demonstrated strong activity in aqueous-phase hydrogenation of biomass-derived oxygenates (xylose, FAL, xylitol, etc.) recently [11-14]. And they are considered as more attractive than noble metal catalysts because of their easy hydrogenation properties and relatively low cost [8]. Nakagawa et al. found that total hydrogenation of FAL to THFOL can be achieved with 94% yield in the gas phase over a highly dispersed Ni/SiO2, and they thought that hydrogenation of FAL to THFOL proceeded in two separate steps: FAL to FOL and FOL toTHFOL [15]. Considering the harsh reaction conditions and undesired byproducts, there is an urgent need to develop modified Ni catalyst, which is applied to FAL hydrogenation in mild reaction conditions. Furthermore, the geometric and electronic properties of supported catalysts are also connected with the supports, influencing the adsorption, desorption and reaction performance of active metals. Activated carbons (AC) are commonly used in catalytic processes, because of their low cost, wide availability, high specific surface area, easily surface modification [16, 17]. Recently, we have demonstrated that Cu/AC-SO3H catalyst, prepared by liquid phase reduction method, was active for liquid-phase hydrogenation and transfer hydrogenation of FAL with 100% selectivity to FOL [18]. Unlike Cu-based catalysts, the adsorption of FAL on Ni occurs through both η1(O)-aldehyde and η2(C, O)-aldehyde modes, resulting in the hydrogenation of C=O and C=C groups over Ni-based catalyst [19]. However, there is still lack in extensive research on selective synthesis of FOL and THFOL from FAL over monometallic Ni catalyst [20], because more or less it is inevitable to generate some unwanted byproducts, like furan, 2-MF and/or 2-methyltetrahydrofuran (2-MTHF) at harsh reaction conditions [15, 21, 22]. To address this issue, in this work, sulfonate group modified active carbon supported Ni catalyst (Ni/AC-SO3H) was successfully developed to selectively transform FAL toward FOL or THFOL by varying reaction parameters. The effects of the sulfonate group on the activity and product selectivity were also investigated in detail.
In a typical experiment, Ni based catalysts were prepared by the liquid phase reduction method, using procedures identical to those in a previous study [18]. Ni/AC and Ni/AC-SO3H were prepared using the same method except Ni(NO3)2·6H2O used instead. The detail information of catalyst preparation and characterization was described in Supporting information.
Catalytic hydrogenation of FAL was carried out in a 25 mL stainless steel autoclave with a magnetic stirrer, a pressure gauge and automatic temperature control apparatus. In a typical experiment, the mixed solutions of FAL, catalyst and 2-propanol were loaded into the reactor. Prior to each run, the Ni-based catalysts were used directly without any pre-reduction treatments. The reactor was sealed, purged three times with N2 at 1.0 MPa, then pressurized with H2 to 4.0 MPa, heated to certain temperature and stirred. After reaction, the autoclave was cooled down quickly. The autoclave contents were transferred to a centrifuge tube, and the used catalyst was separated by centrifugation. The operations before and after the reaction were handled in a glove box. The liquid product was identified and quantitatively analyzed by GC (Shimadzu, GC-2010 Plus) using n-octanol as an internal standard.
The XRD patterns (Fig. S1 in Supporting information) of the catalysts showed broad amorphous features and weak peaks of crystalline phase, consistenting with the results of TEM (Fig. S2 in Supporting information). The FT-IR spectra (Fig. S3 in Supporting information) revealed that -SO3H group were grafted successfully on the surface of active carbon in Ni/AC-SO3H catalyst [23]. Fig. S4a (Supporting information) showed the XPS spectrum of Ni/AC and Ni/AC-SO3H. Typical elements were all detected, and the atomic percentage of S was estimated to be 1.15 at% for Ni/AC-SO3H catalyst. The Ni/AC-SO3H exhibited S 2p peak at 168.9 eV, which was attributable to -SO3H group, in the insert pictures of Fig. S4b in Supporting information [24]. It further indicates that -SO3H group had been grafted successfully on the surface of active carbon in Ni/AC-SO3H catalyst. Ni 2p XPS peaks showed complicated patterns (Fig. S4c in Supporting information). Comparing with unmodified Ni/AC, the Ni/AC-SO3H catalyst showed a little shift in their Ni 2p binding energies from standard value. It could be attributed to an enhanced interaction between Ni species and ACSO3H support [25]. Major Ni species in these two catalysts were oxidation state nickel, and a small amount of metal nickel (Ni) was also found. The acid densities of Ni/AC-SO3H was 0.524 mmol/g (Table S1 in Supporting information). The BET surface area and pore volume deceased after sulfonation, indicating successful grafting of functional groups. Still, the Ni/AC-SO3H catalyst showed a high surface area of 519 m2/g (Table S1 in Supporting information). The S content of the Ni/AC-SO3H was 0.27 mmol/g, which was determined by CHNS elemental analysis (Table S1 in Supporting information). The Ni/AC-SO3H catalyst had higher Ni dispersion (13.17%) and Ni surface area (12.97 m2/g), smaller particle size (7.7 nm) than Ni/AC (9.81%, 9.66 m2/g and 9.3 nm) (Fig. S5 and Table S1 in Supporting information).
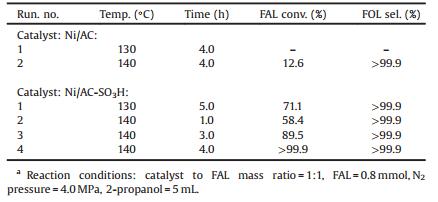
In order to investigate the sulfonate group effect on catalytic performance of Ni based catalysts, we tested the catalytic activity of two kinds of catalysts in the FAL hydrogenation, as shown in Table 1. At very low reaction temperature (60 ℃), the prepared Ni/AC-SO3H catalyst showed very excellent activity (almost 100% FAL conversion and 100% FOL selectivity), but the Ni/AC showed no catalytic activity at the identical reaction condition. With temperature increasing, the Ni/AC-SO3H catalyst showed a shift in product selectivity from FOL to THFOL. Obviously, it exhibited a more promising FAL hydrogenation performance than Ni/AC catalyst in terms of activity and selectivity. The catalytic results revealed that -SO3H group enhanced FAL conversion and THFOL selectivity at moderate and low reaction temperature. Based on Arrhenius law, the corresponding apparent activation energy (Ea) was 56 kJ/mol for Ni/AC-SO3H and 110 kJ/mol for Ni/AC catalyst (Fig. S6 in Supporting information). In addition, the calculated TOF value of Ni/AC-SO3H was larger than Ni/AC (Table S2 in Supporting information), indicating higher reaction rate on Ni/AC-SO3H catalyst. From above characterizations, the Ni/AC-SO3H catalyst displayed better dispersion and smaller particle size of Ni nanoparticles, thus giving a better catalytic performance than that of Ni/AC catalyst. More importantly, comparing with the reported Ni-based catalyst, the as-prepared Ni/AC-SO3H catalyst demonstrated high activity at a more moderate reaction condition, as shown in Table S3 in Supporting information, somehow even be superior to some supported noble catalysts. Besides, the loading amount of Ni was another important factor affecting catalytic performance (Table S4 in Supporting information) [26].
|
|
Table 1 Hydrogenation of FAL over Ni catalysts.a |

To obtain a better understanding of reaction mechanisms and optimize the reaction conditions, series of experiments were conducted over Ni/AC-SO3H catalyst. First, the effect of reaction temperature on selective hydrogenation of FAL were studied in the range of 60–180 ℃ (Fig. S7 in Supporting information). At low reaction temperature (< 70 ℃), FOL is the main product that indicates hydrogenation is prone to take place on the aldehyde group of side chain under this reaction condition. The selectivity of FOL decreased and that of THFOL increased with raising reaction temperature because of further hydrogenation of furan ring. The selectivity of THFOL decreased and that of other by-products (like 2-MF, 2-MTHF) increased when reaction temperature was higher than 140 ℃ because deep hydrogenolysis (C-C or C-O cleavage) occurred inevitably. These results proved the activities of FAL hydrogenation over the Ni/AC-SO3H catalyst is temperature sensitive. It is concluded that 60–70 ℃ and 80–140 ℃ can be considered as optimum reaction temperature ranges for maximum selectivity of FOL and THFOL from the hydrogenation of FAL, respectively. Then, the influences of reaction time on product distribution were observed. As presented in Fig. S8 in Supporting information, it was clearly seen that almost 100% FAL conversion was gained with reaction time prolonged to 3.5 h at the temperature of 100 ℃. Meanwhile, the selectivity of THFOL was raised and that of FOL was decreased gradually, indicating that the THFOL was obtained from the further hydrogenation of furan ring in the intermediate FOL. The effect of H2 pressure was investigated by varying it in the range of 2.0–4.0 MPa, as shown in Table S5 in Supporting information. Obviously, the results revealed that FAL conversion and THFOL selectivity were upgraded with increasing of H2 pressure. In addition, solvent effect also influenced catalytic activity of liquid-phase hydrogenation of FAL and the relevant results were also shown in Table S5 in Supporting information. We found that 68.9% FAL conversion in the non-polar toluene was obtained, which is lower than other protic solvent systems. The selectivity of THFOL showed the following sequence: 2-propanol > water > ethanol. This result may be induced by the difference of solvent properties, such as polarity and solubility, and the relevant solvent polarity sequence: water > ethanol > 2-propanol > toluene [21]. Moreover, 2-propanol had a better solubility of FAL than ethanol and water. So, 2-propanol was found to be an ideal medium which enhanced the catalytic activity. Additionally, the influence of catalyst to FAL mass ratio in the hydrogenation of FAL was also investigated by varying FAL dosage (Fig. S9 in Supporting information).
The roles of Ni species should be investigated to calrify the catalytic model for FAL hydrogenation over Ni/AC-SO3H catalyst. The XPS results showed that major Ni species were oxidation state nickel (NiO, Ni(OH)2 and NiOOH) with a small amount of metal nickel (Ni). For comparison, Ni/AC-SO3H catalyst was reduced by H2 gas at 350 ℃. The XRD patterns confirmed that oxidation state nickel was totally reduced to metallic Ni (Fig. S10 in Supporting information). The FAL hydrogenation over the reduced Ni/AC-SO3H catalyst was also tested (Table S6 in Supporting information). THFOL selectivity of the reduced catalyst was lower than the unreduced Ni/AC-SO3H catalyst, and byproducts (2-MF, 2-MTHF) were produced because of overreaction induced by metallic Ni. The results indirectly proved that oxidation state nickel species play very important roles in reducing the production of byproducts, thus promoting target product selectivity. Zhang et al. concluded that K-doped catalysts enhanced the FAL adsorption and promoted the FAL decarbonylation reaction [27]. Chen and co-workers confirmed that the promoting effect of Mo-dopant was attributed to the acidic property of MoO3, which was favorable for the C=O adsorption in the liquid phase hydrogenation of FAL to FOL [28]. On these bases, we assumed that metallic Ni as active centers can provide the sites for H2 dissociation, while the oxidation state nickel and sulfonate group species are able to adsorb and activate the -C=O bond of FAL molecules, thus greatly improving the hydrogenation performance. In brief, FAL hydrogenation reaction will take place when the active H atom attacks to the bonded aldehyde group by η1 (O)-aldehyde configuration or to the C=C in the furan ring by η2(C, O)-aldehyde configuration by depending on the reaction conditions [15, 22].
Based on the above experimental results, we evaluated the recyclability of Ni/AC-SO3H catalyst at the low conversion. The activity of the catalyst displayed little variation during three loop operations, followed by a slight decrease of THFOL selectivity in the fourth and fifth cycles (Fig. 1). The results suggested the catalyst is reasonably stable even at a high concentration of reactant. The spent catalyst, Ni/AC-SO3H after five recycles, was characterized by ICP, element analysis, TEM, XRD and XPS. The Ni content in the spent catalyst was 14.62 wt%, which is in close proximity to the fresh catalyst (14.78%). The S content of the spent Ni/AC-SO3H was 0.25 mmol/g, which is also similar with it in the fresh catalyst (0.27 mmol/g). Fig. S11 in Supporting information showed the TEM images of the fresh and spent catalyst, which could provide clues concerning the good stability of Ni/AC-SO3H catalyst. The XRD (Fig. S12 in Supporting information) and XPS (Fig. S13 in Supporting information) analysis of the spent catalyst indicated that the Ni/AC-SO3H catalyst is stable. XRD patterns of the spent catalyst revealed oxidation state Ni(OH)2 decreased gradually with the increase of cycling times. It can be inferred indirectly that the catalyst could be reduced to metallic Ni slowly under high H2 pressure.

|
Download:
|
| Fig. 1. Recyclability of Ni/AC-SO3H catalyst in FAL hydrogenation (Reaction conditions: catalyst to FAL mass ratio = 1:4, FAL = 3.2 mmol, reaction temperature = 100 ℃, H2 pressure = 4.0 MPa, 2-propanol = 5 mL, reaction time = 2.0 h.). | |
The employment of liquid organic hydrogen donors alleviates the safety concern of handling high-pressure, flammable hydrogen gas, enhances the solubility of the hydrogen donor in liquid-phase reactions, and substantially reduces the complexity and cost of the experimental setup [29-31]. Here, we investigated the catalytic performance of transfer hydrogenation of FAL by employing Ni/AC-SO3H and Ni/AC as catalysts and 2-propanol as hydrogen donor, the results were shown in Table 2. It could be seen that FOL was the main product derived from FAL, and a certain amount of acetone was also detected, which was derived from 2-propanol dehydrogenation. The experiments clearly showed that at the same reaction condition (140 ℃, 4.0 h), Ni/AC-SO3H, which gained complete conversion, exhibited better transfer hydrogenation performance than Ni/AC (12.6% FAL conversion), indicating -SO3H group could obviously enhance the catalytic performance of transfer hydrogenation, similar with that of H2 hydrogenation. Herein, 2-propanol dehydrogenation required a relatively high temperature, which has been confirmed by many researchers [18, 29, 32]. From Table 2, we concluded that reaction time and reaction temperature were also the two key parameters influencing transfer hydrogenation of FAL. In this work, the Ni/AC-SO3H catalyst gives a coexistence of the Lewis and Brønsted acid sites, which are derived from the nickel oxides and -SO3H groups, respectively. The Lewis acid sites created on metal oxides, which treat as the adsorption and active site, has been widely confirmed by many researchers [33, 34]. Besides, Brønsted acid sites modified metal catalysts also showed good catalytic activities in the selective 1, 3-butadiene hydrogenation and reduction of nitroaromatics [35, 36]. Herein, we proved that the high performance of Ni/AC-SO3H catalyst derives from the synergy between Lewis acid sites (nickel oxides) and Brønsted acid sites (-SO3H). The reaction processes for transfer hydrogenation of FAL were similar with the hydrogenation process (Scheme S1 in Supporting information).
|
|
Table 2 Catalytic transfer hydrogenation of FAL over Ni catalysts.a |
{kind=link}
In summary, we showed that FOL and THFOL can be produced selectively from FAL over Ni/AC-SO3H catalyst, which was prepared by liquid phase reduction method, simply by tuning reaction conditions and the properties of the support. The Ni/AC-SO3H demonstrated a high FOL (< 70 ℃) and THFOL selectivity (>100 ℃) with complete FAL conversion, respectively, and could be reused several times without loss in activity. More importantly, in the transfer hydrogenation of FAL to FOL, an almost 100% FAL conversion was obtained at 140 ℃ after 4.0 h reaction.
AcknowledgmentsWe appreciate the National Natural Science Foundation of China (Nos. 51502297, 51372248, and 51432009), Instrument Developing Project of the Chinese Academy of Sciences (No. yz201421).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2018.03.030.
| [1] |
Y. Roman-Leshkov, C.J. Barrett, Z.Y. Liu, J.A. Dumesic, Nature 447 (2007) 982-985. DOI:10.1038/nature05923 |
| [2] |
P. Gallezot, Chem. Soc. Rev. 41 (2012) 1538-1558. DOI:10.1039/C1CS15147A |
| [3] |
X.M. Liao, V. Pitchon, P.H. Cuong, W. Chu, V. Caps, Chin. Chem. Lett. 28 (2017) 293-296. DOI:10.1016/j.cclet.2016.06.043 |
| [4] |
R. Li, J. Zhao, D. Han, X. Li, Chin. Chem. Lett. 28 (2017) 1330-1335. DOI:10.1016/j.cclet.2017.04.028 |
| [5] |
J.P. Lange, E. van der Heide, J. van Buijtenen, R. Price, ChemSusChem 5 (2012) 150-166. DOI:10.1002/cssc.201100648 |
| [6] |
X. Li, P. Jia, T. Wang, ACS Catal. 6 (2016) 7621-7640. DOI:10.1021/acscatal.6b01838 |
| [7] |
C.P. Jiménez-Gómez, J.A. Cecilia, D. Durán-Martín, et al., J. Catal. 336 (2016) 107-115. DOI:10.1016/j.jcat.2016.01.012 |
| [8] |
K. Yan, G. Wu, T. Lafleur, C. Jarvis, Renew. Sustain. Energy Rev. 38 (2014) 663-676. DOI:10.1016/j.rser.2014.07.003 |
| [9] |
Y. Nakagawa, K. Tomishige, Catal. Commun. 12 (2010) 154-156. DOI:10.1016/j.catcom.2010.09.003 |
| [10] |
Y. Nakagawa, M. Tamura, K. Tomishige, ACS Catal. 3 (2013) 2655-2668. DOI:10.1021/cs400616p |
| [11] |
J. Lee, Y. Xu, G.W. Huber, Appl. Catal. B 140- 141 (2013) 98-107. |
| [12] |
S. Sitthisa, D.E. Resasco, Catal. Lett. 141 (2011) 784-791. DOI:10.1007/s10562-011-0581-7 |
| [13] |
X. Kong, R. Zheng, Y. Zhu, et al., Green Chem. 17 (2015) 2504-2514. DOI:10.1039/C5GC00062A |
| [14] |
A.B. Dongil, L. Pastor-Pérez, A. Sepúlveda-Escribano, R. García, N. Escalona, Fuel 172 (2016) 65-69. DOI:10.1016/j.fuel.2016.01.002 |
| [15] |
Y. Nakagawa, H. Nakazawa, H. Watanabe, K. Tomishige, ChemCatChem 4 (2012) 1791-1797. DOI:10.1002/cctc.v4.11 |
| [16] |
A. Onda, T. Ochi, K. Yanagisawa, Green Chem. 10 (2008) 1033-1037. DOI:10.1039/b808471h |
| [17] |
F.Y. Ye, D.M. Zhang, T. Xue, Y.M. Wang, Y.J. Guan, Green Chem. 16 (2014) 3951-3957. DOI:10.1039/C4GC00972J |
| [18] |
W. Gong, C. Chen, Y. Zhang, et al., ACS Sustain. Chem. Eng. 5 (2017) 2172-2180. DOI:10.1021/acssuschemeng.6b02343 |
| [19] |
S. Sitthisa, W. An, D.E. Resasco, J. Catal. 284 (2011) 90-101. DOI:10.1016/j.jcat.2011.09.005 |
| [20] |
R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sadaba, M.L. Granados, Energy Environ. Sci. 9 (2016) 1144-1189. DOI:10.1039/C5EE02666K |
| [21] |
B. Chen, F. Li, Z. Huang, G. Yuan, Appl. Catal. A 500 (2015) 23-29. DOI:10.1016/j.apcata.2015.05.006 |
| [22] |
T.V. Kotbagi, H.R. Gurav, A.S. Nagpure, S.V. Chilukuri, M.G. Bakker, RSC Adv. 6 (2016) 67662-67668. DOI:10.1039/C6RA14078E |
| [23] |
W.L. Zhang, H.X. Tao, B.H. Zhang, et al., Carbon 49 (2011) 1811-1820. DOI:10.1016/j.carbon.2010.12.050 |
| [24] |
M. Okamura, A. Takagaki, M. Toda, et al., Chem. Mater. 18 (2006) 3039-3045. DOI:10.1021/cm0605623 |
| [25] |
A.B. Dongil, L. Pastor-Pérez, N. Escalona, A. Sepúlveda-Escribano, Carbon 101 (2016) 296-304. DOI:10.1016/j.carbon.2016.01.103 |
| [26] |
M. Zhou, H. Zhu, L. Niu, G. Xiao, R. Xiao, Catal. Lett. 144 (2013) 235-241. |
| [27] |
W. Zhang, Y. Zhu, S. Niu, Y. Li, J. Mol. Catal. A 335 (2011) 71-81. DOI:10.1016/j.molcata.2010.11.016 |
| [28] |
X.F. Chen, H.X. Li, H.S. Luo, M.H. Qiao, Appl. Catal. A 233 (2002) 13-20. DOI:10.1016/S0926-860X(02)00127-8 |
| [29] |
M.J. Gilkey, B. Xu, ACS Catal. 6 (2016) 1420-1436. DOI:10.1021/acscatal.5b02171 |
| [30] |
K. Nakanishi, A. Tanaka, K. Hashimoto, H. Kominami, Chem. Lett. 47 (2017) 254-256. |
| [31] |
T.A. Natsir, T. Hara, N. Ichikuni, S. Shimazu, Chem. Lett. 46 (2017) 1580-1583. DOI:10.1246/cl.170720 |
| [32] |
M.M. Villaverde, T.F. Garetto, A.J. Marchi, Catal. Commun. 58 (2015) 6-10. DOI:10.1016/j.catcom.2014.08.021 |
| [33] |
S. Chen, Z. Shao, Z. Fang, et al., J. Catal. 338 (2016) 38-46. DOI:10.1016/j.jcat.2016.01.030 |
| [34] |
W. Li, G. Fan, L. Yang, F. Li, Catal. Sci. Technol. 6 (2016) 2337-2348. DOI:10.1039/C5CY01084E |
| [35] |
B. Bachiller-Baeza, J. Peña-Bahamonde, E. Castillejos-López, A. Guerrero-Ruiz, I. Rodríguez-Ramos, Catal. Today 249 (2015) 63-71. DOI:10.1016/j.cattod.2014.12.044 |
| [36] |
X. Wang, B. Guan, Y. He, et al., Nanoscale 7 (2015) 3719-3725. DOI:10.1039/C4NR06341D |