 2018, Vol. 29
2018, Vol. 29 



b MoE Key Laboratory of Macromolecule Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, China
Organic fluorescent materials in the solid state have been attracting increasing attention because of their widely applications in many fields such as organic light-emitting diodes, bio-imaging, optical communications, laser devices and so on [1]. As key factors to evaluate the designed materials, the fluorescence color and efficiency are mainly focused on. Generally, the ideal materials could be achieved by molecular structure modification [2, 3]. For example, lowering the π-conjugation benefits the blue emission and the quantum yield. In the contrary, enlarging the π skeleton has adverse effect on the efficiency but is good for the longwavelength fluorescence. Consequently, highly efficient red emissive organic material is relatively rare because of the contradiction between the long-wavelength and the high efficiency. Much effort have been made to resolve this problem [4]. For example, designing three-dimensional molecular structures or modulating the molecules with non-planar arms can effectively eliminate the π-π interactions and then facilitate the target molecule highly efficient in solid state [5]. On the other hand, in recent years, simply changing the molecular conformation or molecular packing structures has been attracting considerable interest in the field of modulating the solid state fluorescence which is demonstrated by a lot of work successively [6]. However, the fluorescence color is usually tuned on a small scale with emission maxima difference (Δλ) less than 100 nm by modulating the molecular packing structure in crystalline state. That is to say, the different packing structures merely fine-tune the fluorescence color and usually is not an efficient regulatory factor to obtain multi-colored fluorescence in obvious contrast. The fundamental cause of the small Δλ color change lies in that the emission region of most molecules mainly depends on the intrinsic molecular structures instead of the packing modes.
To solve this problem and achieve multi-colored fluorescence in sharp contrast based on a single molecule in solid state, excitedstate intramolecular proton transfer (ESIPT)-active molecules which likely show dual emission may be chosen. As shown in Fig. 1a, an intrinsic four-level photocycle (E→E*→K*→K→E) is involved in the ESIPT process. On absorption of light, a fast tautomerization process occurs, leading to either the sole emission of the tautomer or a dual emission if there exists a balance between the E* and K* states. For instance, chemical modification of structurally simple 7-hydroxy-1-indanone successfully balances the E* and K*emission and achieves pure white light emission, as reported by Chou and his colleagues [7]. Different with general emitters, the ESIPT-active molecules are likely more sensitive to the molecular conformation or packing modes [8]. This is probably because the proton transfer rate or the K* energy level is easily affected by small changes in molecular conformation or intermolecular interactions. As a consequence, the balance between the local emission band and the long-wavelength K* emission band of ESIPT molecules in solid state may be easily tuned by preparation condition such as solvent polarity, temperature, pH level. Generally, K* emission of the ESIPT molecules displays large Stokes' shifts, many of which are even over 10, 000 cm-1. Thus the Δλ between E* and K* emission bands is usually very large. Modulating the ratio of the two emission bands in ESIPT-active molecules, single-molecule based multi-colored fluorescence with large Δλ in solid state may be available. Moreover, the relationship between the emission behavior and molecular conformation/ packing structures of ESIPT-active molecules is of great importance on disclosing the reasons that may affect the proton transfer rate. Indeed, polymorph-dependent multi-colored fluorescence based on a single ESIPT-active molecule has been achieved by Araki and co-workers [8a]. However, all the polymorphs display pure K* emission without a very large Δλ. In our previous work recently, the E*& K* white emissive crystal and the pure E* blue emissive crystal based on a single ESIPT-active molecule have been obtained [9]. Nevertheless, multi-colored fluorescence covering pure K* emission, E*& K* emission and pure E* emission, which inevitably is more colorful, significant and has a larger Δλ, is still an unopened field.

|
Download:
|
| Fig. 1. (a) Four level ESIPT process and the keto–enol tautomerism, (b) the excited state of 4MPP and (c) polymorphs of 4MPP displaying E*, K* and E*& K* emissions | |
{kind=link}
On this consideration, a structurally simple molecule named 4- methyl-2-(5-[4-dimethylaminophenyl]-lH-pyrazole-3-yl)phenol (4MPP, Fig. 1b) is employed as a prototype, showing the polymorph-dependent multi-colored fluorescence (Fig. 1c) based on a single ESIPT-active organic small molecule. By different preparation processes, we prepared four crystals based on this molecule and three of them have been successfully analyzed by single-crystal X-ray diffraction. Because of the different K* state, the emission color of these crystals displays huge differences. Furthermore, the crystal structures function well in disclosing the relationship between molecular conformation/packing mode and the emission behavior of ESIPT molecule.
Compound 4MPP was synthesized according to our previous method [9]. A solution of 1, 3-diaryl β-diketones (10 mmol) in 20 mL tetrahydrofuran (THF) was heated to 60 ℃. After addition of hydrazine hydrate (100 mmol), the mixture was heated at 60 ℃ for another 12 h. The solution was evaporated and dried in vacuum to get the crude product as canary yellow solid. Vacuum sublimation in 210 ℃ of the crude product gave rise to pure green emissive solid. Different crystals are prepared as following:
Crystal A: In a 20 mL test tube, 5 mg 4MPP was dissolved in 5 mL CHCl3 or THF, then 10 mL methanol was added without destroying the surface of the CHCl3/THF solution. A few days later, upon evaporation at room temperature, sheet like crystals which were marked as crystal A precipitated out from the concentrated solution.
Crystal B: In a round-bottom flask, a concentrated solution with 4MPP in 20 mL CHCl3 was added 80 mL petroleum ether without destroying the surface of the CHCl3 solution. After 24 h at 0 ℃, large amounts of small crystals (crystal B) precipitated at the CHCl3- petroleum ether interface.
Crystal C: In a 20 mL test tube, 5 mg 4MPP was dissolved in 4 mL CHCl3, and then 0.5 mL CH3COOH was added dropwise. 6 mL petroleum ether was added slowly along the side wall of the test tube. After about 72 h, colorless crystals (crystal C) were found in the side wall of the test tube.
Crystal D: In the preparation process of crystal B, very small amounts of D was obtained.
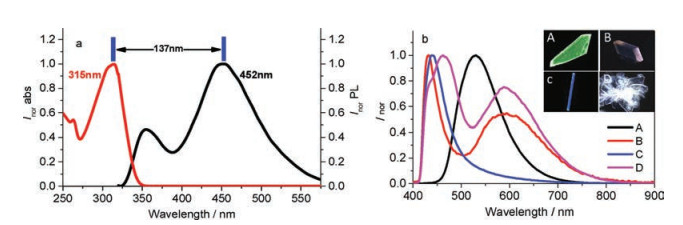
4MPP is well soluble in organic solvent such as CH2Cl2, CHCl3, THF or dimethylsulfoxide (DMSO). As shown in Fig. 2a, 4MPP displays an intense absorption band peaking at 313 nm in CHCl3. 4MPP is weekly emissive in CHCl3 and its emission spectra is featured as a dual emission band which peaking at 355 nm and 452 nm respectively. The weaker emission band corresponds to the E* emission, and the bathochromic-shifted emission peak of 452 nm can be assigned to the K* emission due to the large stokes shifts. The spin-coating film of 4MPP in DMSO or CHCl3 is nonluminous, indicating that the fluorescence quenches in disordered aggregated state. Crystals A–D of the compound show different emission colors (Fig. 2b), the main reason is the difference of K* state. As shown in Fig. S1 (Supporting information), the maximum absorption bands of crystals A–D are in the range of 350–370 nm. Compare with stokes shift of solution, the emission behaviors of crystal A–D could be assigned to the various K* state. Crystal A displays a single green color emission band peaking at 530 nm, which can be ascribed to an absolute K* level emission. This also demonstrates that the inverse intramolecular proton transfer rate k(K*→E*) is far behind the positive proton transfer rate k(E*→K*) when molecules in crystal A excited by UV light. The fluorescence quantum yield of crystal A is 0.26. Crystal B shows typical dual emission peaking at 432 nm and 593 nm which corresponds to local emission and K* emission, respectively. The quantum yield of crystal B is 0.16. Crystal C shows deep blue emission with a sharp peak at 439 nm and a quantum yield of 0.17. Unlike the twodimensional extended sheet-like or slab-like crystals of A or B, crystals of C display one-dimensional needle-like structure and some of them can reach a length of 1 cm. Crystal D displays pure white emission with CIE coordinate of (0.323, 0.293). Its local emission band is centered at 462 nm with a small shoulder at 435 nm. Its K* emission band is centered at 588 nm. Appropriate proportion of the two emission bands endows the crystal D with white light emission.

|
Download:
|
| Fig. 2. (a) UV–vis absorption and emission spectra of 4MPP in CHCl3, (b) emission spectra of crystals A–D. Inset: photographs of crystals A–D | |
{kind=link}
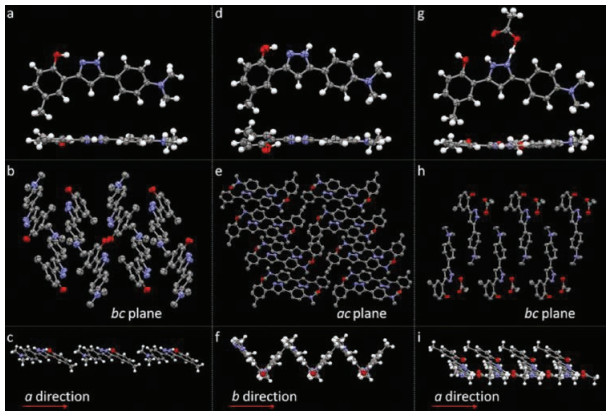
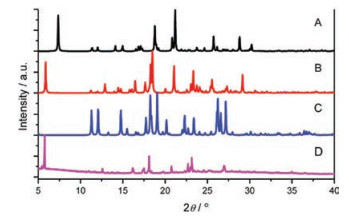
Crystals A–C can be assigned to single-crystal X-ray diffraction. Fig. 3 shows the molecular configuration and packing structures of crystals A–C. The unit cell of crystal A is monoclinic, space group P21/c, containing one molecule. In the individual molecules, the O…N distance between oxygen atom of the hydroxyl group and its neighboring nitrogen atom of the pyrazole ring is 2.579 Å, appropriate for forming a hydrogen bond. Strong intramolecular hydrogen bond O-H…N bridges the hydroxyl group and its neighboring nitrogen atom. The molecules in crystal A display planar molecular conformation with the dihedral angles of 5.51° and 8.09° between the pyrazole ring and adjacent two phenyl rings. Every two molecules connect with each other by intermolecular hydrogen bonds (N-H…O) between the hydroxyl group and pyrazole ring (Fig. S2 in Supporting information). The molecules aggregate into infinite molecular chains in an end-toend packing mode along crystallography a direction, adverse to the π-π stacking and thus beneficial to the fluorescence quantum yield. In the three-dimensional packing structure of crystal A, no π-π interaction exists. The planar molecular conformation and the tightly packing structure without π-overlap, together with the strong intramolecular hydrogen bond, endow the crystals with bright green emission. The unit cell of crystal B is monoclinic, space group P21/n, containing two molecules. The dihedral angles between the pyrazole ring and the two phenyl ring of the two molecules are (22.01°, 7.11°) and (7.03°, 8.97°), respectively. The N…O distances, which relate to how easy (or difficult) it is to form intramolecular hydrogen bond, are 2.663 Å and 2.603 Å, respectively. Every molecule connects with its neighboring two molecules by N-H…π interaction or nonclassical hydrogen bond. Along crystallography b direction, molecules pack into infinite chains in an edge-to-face mode, with the dihedral angle of about 76.50°. Every two molecules assemble into a slip-packing unit along crystallography a direction. And molecules display slipped packing structure along b direction. These packing structures in crystal B effectively prevent the molecules from π-π stacking. In crystal C, the unit cell is orthorhombic, space group P212121, containing one molecule which connects with an acetic acid molecule. The nitrogen atom (N1) close to the p-(dimethylamino) phenyl group share one hydrogen atom with the acetic acid molecule by an extremely strong intermolecular hydrogen bond. The previous hydrogen atom on this nitrogen atom resonates to the other nitrogen atom (N2) which is close to the hydroxyl group. Consequently, intramolecular hydrogen bond between the hydroxyl group and N2 cannot form any more. That is to say, ESIPT process would not happen in crystal C. Molecules in crystal C display planar conformation with the dihedral angles between pyrazole ring and two phenyl rings of 6.86° and 3.67°. Taking the pyrazole molecule and the connected acetic acid molecule as a whole, it is easy to found that, each pair of molecules connect with its neighboring three molecular pairs by C-H…π interactions and intermolecular hydrogen bond. Along crystallography a and b direction, molecules in crystal C display slip-packing modes with different slope and spacing (Fig. S3 in Supporting information). Along crystallography c direction, molecules display an interesting helical packing structure. No π-π interaction is observed in the crystal packing structure of C. Crystal D is obtained in very small amount and the crystal quality is not high enough to be subjected to the single-crystal X-ray diffraction. Instead, single crystal powder X-ray diffraction was measured for D. As shown in Fig. 4, crystal D displays different diffraction signals which also differ from those simulated signals of single crystals A–C, demonstrating that crystal D must have different new packing structures. This also explains why they show different emission behaviors compared with crystals A–C.

|
Download:
|
| Fig. 3. Molecular structures and conformations of (a) crystal A, (d) crystal B and (g) crystal C with thermal ellipsoids at 50% probability, and the packing structures of (b) (c) crystal A, (e) (f) crystal B and (h) (i) crystal C (H-atom are omitted for clarity) | |
{kind=link}

|
Download:
|
| Fig. 4. Powder X-ray diffraction patterns of crystals A–D | |
{kind=link}
Considering the different crystal structures and the distinctively different emission behaviors of crystals A–D, a conclusion is drawn that the emission behavior of ESIPT molecules is sensitive to the crystal structures and can be tuned by small changes of the molecular conformations, packing structures and inter/intramolecular interactions. Among crystals A–D, A displays totally K* emission which is most likely because of the shortest O…N distance which benefits to the proton transfer process; B displays obviously different emission spectra because of their distinction in the molecular conformation and intermolecular interaction compared with A; C displays totally local emission (also known as E* emission) because of the formation of intermolecular hydrogen bond which inhibits the intramolecular hydrogen bond; D display balanced E* and K* emission bands with different proportion because of their different packing modes compared with the other three crystals and successfully achieve white emission. Overall, small difference of crystal structure may result in big change of ESIPT molecules' emission behavior. As a consequence, multi-colored fluorescence in a large region based on a single molecule is available.
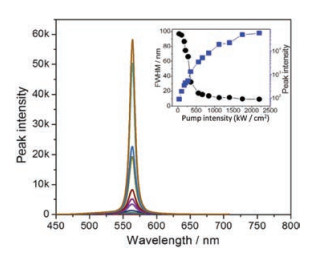
When a slice of the single crystal of crystal A is excited by a 355 nm picoseconds pulse of elliptic excitation area, the green emission is amplified into the intense and narrow green amplified spontaneous emission band. As the intensity of the pump beam emited on the sample is turned up from 35 kW/cm2 to 1750 kW/cm2, the FWMH gradually decreases from 96.9 nm to 9.7 nm (Fig. 5). The threshold value is about 153 kW/cm2 determined by the slope mutation of the curve depicting the full width at half maximum (FWHM) as a function of the laser pump intensity. The gain coefficient is measured by using a slit in front of the laser beam. As shown in Fig. S4 (Supporting information), there is an exponential growth in the emission intensity on increasing the width of the slit. And the maximum gain coefficient value is 46.57 cm-1.

|
Download:
|
| Fig. 5. PL spectra as a function of the pump laser energy (inset: dependences of emission peak intensity and FWHM of emission spectra) | |
{kind=link}
In summary, a structurally simple 4MPP molecule is synthesized as a prototype, showing the multi-color fluorescence over large-area tunable emission region. Changing the preparation condition, crystals A–D which show differentproportion characteristic emission bands are obtained. These crystals display distinctively different emission which covers deep blue, bright green, white and pink white region. The crystals structures clearly give evidence that the fluorescence of ESIPTactive molecules is very sensitive to the changes in crystal structure and thus are changeful and easily tunable. Furthermore, the crystal structures of the multiple emissive crystals would play important roles in disclosing the relationship between molecular conformation/packing mode and the proton transfer rate in ESIPT molecules
AcknowledgmentThis work was supported by the National Natural Science Foundation of China (Nos. 51622304 and 51773077).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2018.08.003.
| [1] |
(a) D. Li, H. Zhang, Y. Wang, Chem. Soc. Rev. 42(2013) 8416-8433; (b)Y. Xua, M. Tian, H. Zhang, et al., Chin. Chem. Lett. 29(2018) 1093-1097; (c)Y. Luo, Z. Lu, Y. Huang, Chin. Chem. Lett. 27(2016) 1223-1230; (d)M. Bio, P. Rajaputra, G. Nkepang, Y. You, J. Med. Chem. 57(2014) 3401-3409; (e)H. Mizuno, I. Ohnishi, H. Yanagi, F. Sasaki, S. Hotta, Adv. Mater. 24(2012) 2404-2408; (f)H. Liu, Y. Li, Q. Gao, Z. Liu, H. Fu, Chin. Chem. Lett. 29(2018) 209-212; (g)B. Tang, Z. Zhang, H. Liu, H. Zhang, Chin. Chem. Lett. 28(2017) 2129-2132; (h)P. Audebert, F. Miomandre, Chem. Sci. 4(2013) 575-584; (i)K. Nagura, S. Saito, H. Yusa, et al., J. Am. Chem. Soc. 135(2013) 10322-10325; (k)L. Wang, K. Ye, H. Zhang, Chin. Chem. Lett. 27(2016) 1367-1375. |
| [2] |
(a) B.K. An, S.H. Gihm, J.W. Chung, et al., J. Am. Chem. Soc. 131(2009) 3950-3957; (b)P. Biegger, S. Stolz, S.N. Intorp, et al., J. Org. Chem. 80(2015) 582-589. |
| [3] |
(a) M. Shimizu, R. Kaki, Y. Takeda, et al., Angew. Chem. Int. Ed. 51(2012) 4095-4099; (b)S.H. Liao, J.R. Shiu, S.W. Liu, et al., J. Am. Chem. Soc. 131(2009) 763-777. |
| [4] |
(a) Y. Tao, Q. Wang, C. Yang, et al., Angew. Chem. Int. Ed. 120(2008) 8224-8227; (b)D.H. Kim, N.S. Cho, H.Y. Oh, et al., Adv. Mater. 23(2011) 2721-2726; (c)H.H. Chou, C.H. Cheng, Adv. Mater. 22(2010) 2468-2471; (d)H. Fukagawa, T. Shimizu, H. Hanashima, et al., Adv. Mater. 24(2012) 5099-5103; (e)Y. Cho, J. Lee, Adv. Mater. 23(2011) 4568-4572. |
| [5] |
C. Borek, K. Hanson, P.I. Djurovich, et al., Angew. Chem. 119 (2007) 1127-1130. DOI:10.1002/(ISSN)1521-3757 |
| [6] |
(a) K. Wang, H. Zhang, S. Chen, et al., Adv. Mater. 26(2014) 6168-6173; (b)X. Cheng, F. Li, S. Han, et al., Sci. Rep. 5(2015) 9140; (c) T. Seki, S. Kurenuma, H. Ito, Chem.-Eur. J. 19(2013) 16214-16220; (d)T. Shida, T. Mutai, K. Araki, CrystEngComm 15(2013) 10179-10182; (e) E.Q. Procopio, M. Mauro, M. Panigati, et al., J. Am. Chem. Soc. 132(2010) 14397-14399; (f)M.A. Malwitz, S.H. Lim, R.L. White-Morris, et al., J. Am. Chem. Soc. 134(2012) 10885-10893. |
| [7] |
K.C. Tang, M.J. Chang, T.Y. Lin, et al., J. Am. Chem. Soc. 133 (2011) 17738-17745. DOI:10.1021/ja2062693 |
| [8] |
(a) T. Mutai, H. Tomoda, T. Ohkawa, Y. Yabe, K. Araki, Angew. Chem. Int. Ed. 47(2008) 9522-9524; (b)R. Wei, P. Song, A. Tong, J. Phys. Chem. C 117(2013) 3467-3474. |
| [9] |
H. Liu, X. Cheng, H. Zhang, Y. Wang, S. Yamaguchi, Chem. Commun. 53 (2017) 7832-7835. DOI:10.1039/C7CC03758A |