 2018, Vol. 29
2018, Vol. 29 



b High Magnetic Field Laboratory, Chinese Academy of Sciences, Hefei 230031, China
Fluorescence spectroscopy, either steady-state or time-resolved modes, is more and more widely utilized for in vivo imaging and especially for the studies of localization, motility and structure of proteins in living cells [1-3]. The time-dependent fluorescence decays, known as the fluorescence lifetime, is based on the time duration that the fluorescence molecule stays in its excited state before emitting a photon [4]. The measurement of the fluorescence lifetime depends directly on excited-state reactions, which is affected by several factors, including the presence of neighboring molecules, the dielectric constant of the medium, temperature, and pressure [5]. Therefore, the fluorescence lifetime is frequently used to monitor local environmental perturbations to uncover details about the conformations and dynamics of macromolecules [6, 7]. Considering the fact that the fluorescence lifetime is independent of chromophore concentration, and avoids the effects of photon scattering or bleaching in live cells, this method is very much suitable for studies with low fluorophore concentrations, such as investigations of membrane proteins. The fluorescence lifetime imaging microscopy (FLIM) is well developed to be an imaging technique with spatial variations of fluorescence lifetimes, rather than intensities of fluorophores [2, 8]. Combining with the cutting-edge photon-counting detection methods, FLIM is thus an ideal tool to track the conformations and dynamics of fluorescence-marked membrane proteins in pharmacology studies.
Ferroportin (FPN), the sole iron exporter known in vertebrates to date, plays an important role in maintaining cellular iron homeostasis [9]. It is located on the surface of intestinal enterocytes, macrophages, hepatocytes, and placenta where it releases cellular iron into the plasma, regulating its absorption and recycling [10-12]. The FPN is regulated at several levels including presumed transcriptional regulation, translational regulation, and post-translational regulation by hepcidin [13] and iron [14, 15]. Besides iron, FPN can also mediate export of other essential metal ions, such as Mn2+ [16] and Zn2+ [17]. As another essential trace element, Mn2+ plays essential roles in human development and multiple physiological functions [18]. The way that accumulates Mn2+ is similar as iron [19, 20], and the extracellularly transporting of Mn2+ is essential for regulation of cytoplasm metal ion level. Deficiency of FPN was reported to result in Mn2+ metabolism abnormal in mice [21]. Mn2+ exposure can increase the expression level of FPN in HEK293T cells as well as in mouse cerebellum and cortex [22, 23]. Although all of these studies suggested FPN as an exporter of Mn2+ and implied that FPN expression may be responsible for Mn2+ concentrations, no studies have addressed how the extracellular Mn2+ affect FPN from outside of mammalian cells.
Recently, a homology model of human FPN was build based on the only known crystal structure of a putative bacterial transitionmetal transporter BbFPN, which showed outward- and inwardfacing structures during transportation cycle, while the flexible intracellular side of the N lobe is fitting to that of the C lobe, and transiting the conformation from inward- to outward-facing states [24]. Therefore, it is wondered whether there existed two conformations in cell membrane during Mn2+ transport cycle.
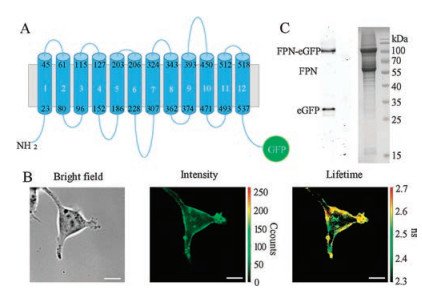
Herein, the fluorescence lifetime measurement was applied to analyze the FPN conformation states upon the treatment of Mn2+. We initiated with constructing a plasmid with eGFP fused in C terminal of FPN (FPN-eGFP) [25] (Fig. 1A). Then the FPN-eGFP construct was expressed in HEK293T cells and analyzed using FLIM system. Both fluorescence intensity and lifetime based imaging demonstrated clear membrane expression of FPN-eGFP (Fig. 1B). To ensure the green fluorescence observed here was from fusing protein, the membrane fraction was collected after harvesting the HEK293F expressing FPN-eGFP, and the SDS-PAGE analysis using both Coomassie brilliant blue staining and eGFP exposure clearly showed the successful FPN-eGFP expression, despite of some selfcleaved eGFP tags (Fig. 1C). At the same time, the fluorescence imaging of HEK293T cells co-transfected with eGFP plasmid or eGFP plasmid alone showed green fluorescence mostly in cytoplasm Figs. 2A and B), indicating the membrane expression of FPN-eGFP. The slight green fluorescence in cytoplasm might due to some broken eGFP showed in fluorescence imaging (Fig. 1C).

|
Download:
|
| Fig. 1. (A) Topology of FPN-eGFP, with eGFP attached at C-terminal of FPN. (B) Membrane expression of FPN-eGFP in HEK293T cell, as shown in bright field (left), intensity (middle) and lifetime (right). Scale bar: 10 mm. (C) Coommassie-stained SDS-PAGE gel (right) and the fluorescence detection (left) of the extracted FPN-eGFP protein from cell membrane | |
{kind=link}

|
Download:
|
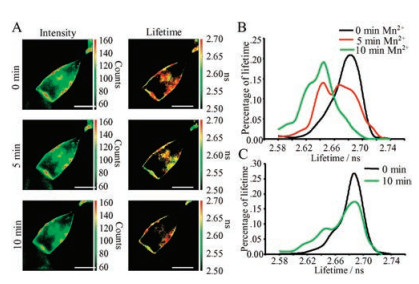
| Fig. 2. Fluorescence lifetime analysis of selected pixels on HEK293T cell membrane. The fluorescence intensity imaging of HEK293T cells with expression of FPN-eGFP (A), eGFP (B) FPN + eGFP (co-expression of FPN and eGFP) (C). The white square showed the selected area for fluorescence lifetime imaging, which was fitted using multi-component exponential decays, before (middle) and 10 min after (right) treatments of 0.5 mmol/L MnCl2. τ is the calculated lifetime result and χ2 represents the fitting quality (χ2 < 1.2 indicates valid lifetime fitting). (D) Scattering dots showed the lifetime change (Y axis) versus intensity change (X axis) of each FLIM pixel in a single cell expressing FPN-eGFP before and after 0.5 mmol/L MnCl2 treatments | |
{kind=link}
The FLIM system (Q2, ISS) allowed us to choose single pixel on cell membrane imaging for further lifetime analysis [26]. A 488- nm diode-pulsed laser and a picosecond photon counting detector were applied. To detect the influence of Mn2+ on FPN proteins expressed on cell membrane, we replaced the solution from the conventional saline to the 0.5 mmol/L Mn2+-containing solution, while not using local perfusion, to avoid the interference of flow turbulence. After 5 min or 10 min 0.5 mmol/L Mn2+ treatment, the fluorescence imaging of the single cell with lifetime or intensity data was collected. Several single-pixels were chosen on the membrane of a HEK293T cell expressing FPN-eGFP, to obtain fluorescence lifetime values through intensity decays fitting with a sum of multiple exponential components using the software (Vistavision, ISS). The same pixel on cell membrane was chosen and analyzed after 10 min Mn2+ treatment (Fig. 2C).
Since the fluorescence lifetime is sensitive to the microenvironment, the observed changes of lifetime value are good indicators for the conformation changes or local environment variations surrounding the fluorophore. Here, the decreased lifetime value of the FLIM data of FPN-eGFP upon Mn2+ treatment suggested that eGFP was in a situation with high probability of quenching. Considering the mobilization of cell membrane lipid, which has constant variation and fluctuation of membrane proteins (integral or peripheral) components or amounts in each FLIM pixel, we generated the correlation plots with fluorescence lifetime versus intensity for every FLIM pixel (Fig. 2D). The change of lifetime or intensity was calculated as the initial value minus the value after 10 min treatment. It clearly showed that the intensity changes ranged from -200 to +200 counts, which may due to the random motion of membrane protein into or out of each pixel. At the same time, the lifetime changes were mostly above positive axis, suggesting that the decreased fluorescence lifetime values in FLIM images were not due to the motion of membrane proteins but the conformational changes of FPN-eGFP upon 10 min Mn2+ treatment. HEK293T cells expressing with eGFP alone (Fig. 2A) or FPN + eGFP (Fig. 2B) were also measured and analyzed in similar way. The derived fluorescence lifetime values showed no obvious changes from this FLIM pixel. The scatter dots distribution were observed around zero, suggesting that Mn2+ treatment did not affect the eGFP lifetime in HEK293T cells (Fig. 2D).
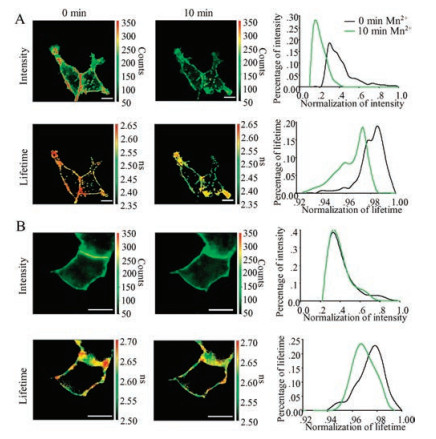
We have collected series of fluorescence intensity and lifetime imaging of a HEK293T cell expressing FPN-eGFP upon Mn2+ treatment (Fig. 3A), demonstrating a clear down-regulation of lifetime as the color changed from hot (larger lifetime) to cold (smaller lifetime). Since there are at least two conformational states reported in previous crystal structure studies [24], we prefer not to use the averaged lifetime of the whole cell but analyzed the population distributions with different lifetime values for each FLIM pixel (Fig. 3B), to illustrate existence of distinctive conformational states. In the distribution curve (Fig. 3B), lifetime values before Mn2+ treatment showed a major peak, suggesting that most FPN-eGFP proteins stay in a specific conformation, which could be represented by this peak. The lifetime distribution peaks shifted the curve to the left after 10 min Mn2+ treatment and showed another large peak with a shorter lifetime, which might represent another conformation state. A shorter time Mn2+ treatment (5 min) showed two peaks in the curve with almost equal intensity, representing a probable conformational state inter-change. A negative control was also analyzed, the normal saline buffer without presence of Mn2+ was applied to treat the cells for 10 min, no obvious fluorescence lifetime distribution changes of FPN-eGFP were observed (Fig. 3C), strongly indicated that the observed fluorescence lifetime distribution changes in Fig. 3B was due to conformational responses upon treatment of Mn2+. The data suggested that there might exist two FPN conformations before or after Mn2+ treatments, and the Mn2+ treatments of binding to FPN might lead to a pronounced FPN conformation change, with the C-terminal attached eGFP easily quenched by ambient charge ions.

|
Download:
|
| Fig. 3. Fluorescence distribution analysis of HEK293T cells expressing FPN-eGGP with exposure of 0.5 mmol/L MnCl2. (A) The fluorescence intensity and lifetime imaging of the cells before or after 0.5 mmol/L MnCl2 treatment. (B) Fluorescence lifetime distribution of data from panel A (FPN-eGFP in the HEK293T cells). (C) Fluorescence lifetime distribution of FPN-eGFP in HEK293T cells with saline alone (no Mn2+) treatment for 10 min | |
{kind=link}
At the same time, fluorescence lifetime distribution analysis of HEK293T cells transfected with eGFP alone (Fig. S1A in Supporting information) or co-transfected with eGFP and FPN (FPN + eGFP) (Fig. S1B in Supporting information) were collected and analyzed. Obviously, 10 min Mn2+ treatment did not drive the peak shifting in fluorescence lifetime distribution curve, furtherly validating that the fluorescence lifetime shift in HEK293T cells with expression of FPN-eGFP resulted from the FPN conformation changes upon Mn2+ treatments.
A pharmacology blockbuster drug hepcidin was reported to induce FPN internalization, which was observed to result in decreased fluorescence intensity on cell membrane [13]. However, fluorescence intensity decrease could be due to several factors, including fluorescent bleaching and light scattering. To analyze the direct interference of the fluorescent bleaching to our FLIM image analysis, we herein mimicked the decreased fluorescence intensity through fluorescent bleaching using excitation laser with different power. At first, 30%–35% of maximal laser power was set to bleach the fluorescence intensity. After 10 min treatment of 0.5 mmol/L Mn2+, the fluorescence intensity reduced dramatically (Fig. 4A). We then attenuate the laser power at 15%–20%, no obvious peak shifting observed in fluorescence intensity distribution curve, before or after 10 min Mn2+ treatment (Fig. 4B). Interestingly, similar patterns of peak shifting were observed in the fluorescence lifetime distribution curves with either high or low power laser excitation. Therefore, the fluorescence lifetime distribution analysis will be much superior to the fluorescence intensity distribution analysis for in cell protein conformation change analysis, especially upon treatment of external stimulus. That is, the laser power could strongly influence fluorescence intensity analysis while fluorescence lifetime data can sustain to high laser power and be applied to analyze the local environment changes or protein conformation changes around the fluorophore, in high sensitivity.

|
Download:
|
| Fig. 4. Different influence of laser power on FPN-eGFP fluorescence intensity and lifetime. (A) The fluorescence intensity and lifetime imaging of HEK293T cells with expression of FPN-eGFP before and after 10 min 0.5 mmol/L MnCl2 treatment using 30%–35% laser power. The fluorescence distribution of either intensity or lifetime was shown on the right. The strong laser bleached the FPN-eGFP fluorescence intensity and left shifting the lifetime distribution. (B) The fluorescence intensity and lifetime imaging of HEK293T cells with expression of FPN-eGFP before and after 10 min 0.5 mmol/L MnCl2 treatment using 15%–20% laser power. The fluorescence distributions of either intensity or lifetime were shown on the right. The FPN-eGFP fluorescence intensity showed no change but the fluorescence lifetime distribution shift to the left | |
{kind=link}
In conclusion, the fluorescence lifetime imaging microscopy (FLIM) method was applied to analyze FPN conformation changes in a single cell, with observing the fluorescence lifetime or intensity values of eGFP, attached to the C-terminal of FPN (FPNeGFP). The fluorescence lifetime distribution revealed two peaks upon Mn2+ treatment, which is consistent with two major conformation states reported in previous crystal structure studies. This method provided a reliable way to discrete the conformation subsets of membrane proteins in a living cell. Especially, the in cell FLIM analysis could be applied for further studies of hepcidininduced FPN conformation changes in FPN internalization and degradation which is very biological and pharmacological essential, but hard to detect using fluorescence intensity based methods.
AcknowledgmentsThis work was supported by the National Key R&D Program of China (Nos. 2016YFA0400900, 2017YFA0505300), and the Instrument Developing Project of the Chinese Academy of Sciences (No. YZ201564).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2018.04.026.
| [1] |
B.A. Griffin, S.R. Adams, R.Y. Tsien, Science 281 (1998) 269-272. DOI:10.1126/science.281.5374.269 |
| [2] |
P.I. Bastiaens, A. Squire, Trends Cell Biol. 9 (1999) 48-52. DOI:10.1016/S0962-8924(98)01410-X |
| [3] |
Q. Zhang, X. Dong, K.P. Wang, et al., Chin. Chem. Lett. 28 (2017) 777-781. DOI:10.1016/j.cclet.2017.03.001 |
| [4] |
S. Liu, P. Lv, D. Li, et al., Chem. Commun. (Camb.) 51 (2015) 15971-15974. DOI:10.1039/C5CC06124E |
| [5] |
P. Ning, W.J. Wang, M. Chen, Y. Feng, X.M. Meng, Chin. Chem. Lett. 28 (2017) 1943-1951. DOI:10.1016/j.cclet.2017.09.026 |
| [6] |
J.M. Beechem, L. Brand, Annu. Rev. Biochem. 54 (1985) 43-71. DOI:10.1146/annurev.bi.54.070185.000355 |
| [7] |
T. Ha, A.Y. Ting, J. Liang, et al., Proc. Natl. Acad. Sci. U. S. A. 96 (1999) 893-898. DOI:10.1073/pnas.96.3.893 |
| [8] |
T.W.J. Gadella, T.M. Jovin, R.M. Clegg, Biophys. Chem. 48 (1993) 221-239. DOI:10.1016/0301-4622(93)85012-7 |
| [9] |
A. Donovan, A. Brownlie, Y. Zhou, et al., Nature 403 (2000) 776-781. DOI:10.1038/35001596 |
| [10] |
G. Hetet, I. Devaux, N. Soufir, B. Grandchamp, C. Beaumont, Blood 102 (2003) 1904-1910. DOI:10.1182/blood-2003-02-0439 |
| [11] |
A. Donovan, C.A. Lima, J.L. Pinkus, et al., Cell. Metab. 1 (2005) 191-200. DOI:10.1016/j.cmet.2005.01.003 |
| [12] |
H. Zoller, R.O. Koch, I. Theurl, et al., Gastroenterology 120 (2001) 1412-1419. DOI:10.1053/gast.2001.24033 |
| [13] |
E. Nemeth, M.S. Tuttle, J. Powelson, et al., Science 306 (2004) 2090-2093. DOI:10.1126/science.1104742 |
| [14] |
C. Delaby, N. Pilard, A.S. Goncalves, C. Beaumont, F. Canonne-Hergaux, Blood 106 (2005) 3979-3984. DOI:10.1182/blood-2005-06-2398 |
| [15] |
D.L. Zhang, T. Senecal, M.C. Ghosh, et al., Blood 118 (2011) 2868-2877. DOI:10.1182/blood-2011-01-330241 |
| [16] |
M.S. Madejczyk, N. Ballatori, Biochim. Biophys. Acta 1818 (2012) 651-657. DOI:10.1016/j.bbamem.2011.12.002 |
| [17] |
C.J. Mitchell, A. Shawki, T. Ganz, E. Nemeth, B. Mackenzie, Am. J. Physiol. Cell Physiol. 306 (2014) C450-459. DOI:10.1152/ajpcell.00348.2013 |
| [18] |
K.M. Erikson, M. Aschner, Neurochem. Int. 43 (2003) 475-480. DOI:10.1016/S0197-0186(03)00037-8 |
| [19] |
H. Gunshin, B. Mackenzie, U.V. Berger, et al., Nature 388 (1997) 482-488. DOI:10.1038/41343 |
| [20] |
E.A. Malecki, B.M. Cook, A.G. Devenyi, J.L. Beard, J.R. Connor, J. Neurol. Sci. 170 (1999) 112-118. DOI:10.1016/S0022-510X(99)00203-8 |
| [21] |
Y.A. Seo, M. Wessling-Resnick, FASEB J. 29 (2015) 2726-2733. DOI:10.1096/fj.14-262592 |
| [22] |
Z. Yin, H. Jiang, E.S. Lee, et al., J. Neurochem. 112 (2010) 1190-1198. DOI:10.1111/jnc.2010.112.issue-5 |
| [23] |
E.A. Smith, P. Newland, K.G. Bestwick, N. Ahmed, Elem. Med. Biol. 27 (2013) 65-69. DOI:10.1016/j.jtemb.2012.07.002 |
| [24] |
R. Taniguchi, H.E. Kato, J. Font, et al., Nat. Commun. 6 (2015) 8545. DOI:10.1038/ncomms9545 |
| [25] |
A. Periasamy, M. Elangovan, E. Elliott, D.L. Brautigan, Methods Mol. Biol. 183 (2002) 89-100. |
| [26] |
H.C. Gerritsen, M.A. Asselbergs, A.V. Agronskaia, W.G. van Sark, J. Microsc. 206 (2002) 218-224. DOI:10.1046/j.1365-2818.2002.01031.x |