 2018, Vol. 29
2018, Vol. 29 



b Shanghai Key Laboratory of Chemical Biology, School of Pharmacy, East China University of Science and Technology, Shanghai 200237, China;
c Guangxi Scientific Research Center of Traditional Chinese Medicine, Guangxi University of Chinese Medicine, Nanning 530200, China;
d State Key Laboratory of the Discovery and Development of Novel Pesticide, Shenyang Sinochem Agrochemicals R & D Co., Ltd., Shenyang 110021, China
Release of nitric oxide (NO) in a spatially or temporally controlled fashion facilitates investigation of the mechanism of NO in biological processes [1] and can also be harnessed as therapeutic or biomanipulative applications [2, 3]. Compared to various chemo-triggers, e.g., enzymes [4], reducing agents [5], oxidative species [6] and nucleophiles [7], light is particularly desirable due to its versatility, non-invasiveness, and feasibility for biological settings. The flux and the dose of NO are critical to its biological outcome [8]. Modulating the intensity of the photoirradiation may offer a convenient way to adjust the photodecomposition rate of our photo-triggered and photo-calibrated NO donors. However, in cases where the light intensity is not readily adjusted, NO donors of varying releasing kinetics could be a desirable alternative.
The existing photo-triggered NO donors include photo-caged spontaneous NO donors [9], ortho-substituted nitrobenzene [10], and metal-nitroso complexes [11]. One limitation of the aforementioned photo-triggered NO donors is the lack of a mechanism to calibrate the dose and kinetics of NO release.CommercialNOprobes were employed to monitor the NO generation characteristics. However, they detect NO by scavenging NO and therefore prevent NO from eliciting its biological functions. We have recently devised a series of NO donors, which are N-nitrosated push-pull fluorophores (Fig. 1) and addressed this dilemma [12-14]. Their release of NO is not only triggered by light, but also calibrated by simultaneous release of a fluorophore, whose fluorescence signifies the release of NO in a real-time fashion. In particular, NOD560 is interesting because it can be photo-activated by a wide-selection of laser lines, in the spectral range of 375–556 nm and the resulting rhodamine dye is a superior fluorophore for imaging-based applications [14-19]. The only drawback is its relatively slow kinetics. Therefore, development of such NO donors with accelerated NO release rate is an important research topic.

|
Download:
|
| Fig. 1. (A) The resonance structures and NO-releasing mechanism of N-nitrosated push-pull dyes. (B) Three representative NO donors of this class. | |
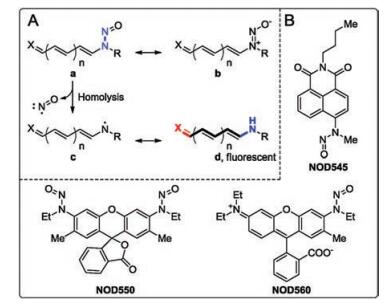{kind=link}
N-Nitrosated secondary amines have been sketched in its N-nitroso form (a, Fig. 1) following the convention. However, we note that the diazonium oxide (b, Fig. 1) is in fact the major contributing resonance structure for N-nitrosamine. Upon photoexcitation of the structure b, the intramolecular charge transfer occurs and the molecule adopts the structure a. The single bond between the two nitrogen atoms (highlighted in blue) in the form a homolyzes to release NO and an anilinyl radical. Based on the above analysis, it becomes obvious that a weak N-N bond is the key to high homolysis tendency. In NOD545 [12], NOD550 [13], and NOD560 [14], the nitrosamine moiety and the chromophore are nearly orthogonal to each. Reducing the dihedral angle in between is expected to effectively promote the electronic delocalization from the nitrogen atom to the dye scaffold, weaken the N-N bond and hence enhance NO release kinetics. Therefore, we designed a novel NO donor with the nitrosamine moiety locked coplanar with the dye scaffold.
2-(4-(Diethylamino)-2-hydroxybenzoyl)benzoic acid (1) was condensed with 1, 2, 3, 4-tetrahydroquinolin-7-ol (2) in concentrated H2SO4 at 100 ℃ for 24 h to furnish the desired rhodamine dye (3) in a 91% yield. Compound 3 was then nitrosated to afford NOD575 in a 94% yield. X-ray diffraction reveals that NOD575 is in its lactone form in solid state. The dihedral angle between the nitrosamine and the rhodamine dye scaffold was measured to be of 12.66°.
The UV–vis absorption and fluorescence properties of both NO575 and 3 were acquired in neutral phosphate buffer with 1% DMSO as co-solvent. The maximal absorption of NOD575 is at 510 nm with two shoulder peaks at 480 nm and 545 nm, respectively, suggesting NOD575 exists predominantly in its ring-open form (Fig. 2). NOD575 is not fluorescent. In comparison, the potential decomposition product is highly absorbing at 545 nm and strongly fluorescent with an emission maximum at 575 nm (Φ = 0.58).

|
Download:
|
| Fig. 2. (A) Dihedral angles between the nitrosamine (orange) and the dye scaffold (green). (B) The synthesis and photo-decomposition of NOD575. | |
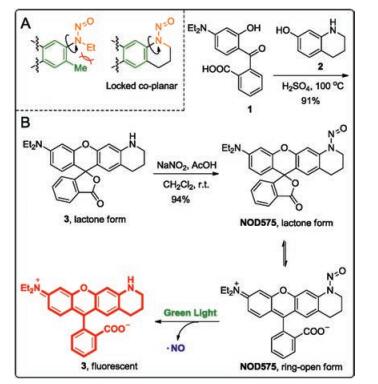{kind=link}
The solution of NOD575 (10 μmol/L) in phosphate buffer (50 mmol/L, pH 7.4) with 1% DMSO was photoirradiated with a laser at 532 nm. The UV–vis absorption and fluorescence emission spectra were recorded intermittently. The absorbance band of NOD575 gradually decreases with respect to duration of photoirradiation, the absorption and emission of the product increases concomitantly. The decomposition product was unambiguously confirmed to be 3, by NMR and MS. It took ca. 100 s for its decomposition to complete, compared to ca. 2300s of NOD560. NOD575 expectedly exhibits a much improved decomposition kinetics compared to NOD560. This confirms our original hypothesis that reducing dihedral angle can weaken N-N single bond and facilitates hemolysis (Figs. 3 and 4).

|
Download:
|
| Fig. 3. The two different views of the crystal structure of NOD575. | |
{kind=link}

|
Download:
|
| Fig. 4. (A) UV–vis absorption spectral and fluorescence emission spectral changes of NOD575 solutions upon photo-irradiation by 532 nm. (B) The enhancement of emission intensity at 575 nm of NOD575 solution with respect to the duration of photo-irradiation. | |
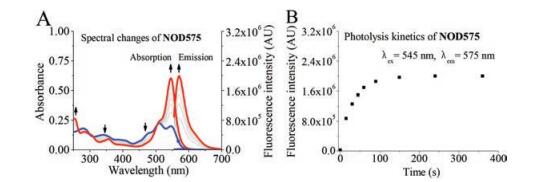{kind=link}
Denitrosation of N-nitrosamines may occur in reductive environment [20]. We have also previously discovered that reducing agents may accelerate the decomposition of NO donors. Therefore, the chemostability of NOD575 (10 μmol/L) was tested by incubation with biological thiols and bio-relevant reducing agents, i.e., cysteine, glutathione, and ascorbic acid, in phosphate buffer (50 mmol/L at pH 7.4) with 1% DMSO in the dark at room temperature. The absorbance of each solution at 545 nm remained unchanged in 24 h, suggesting NOD575 are sufficiently chemostable for practical biological applications (Fig. S3 in Supporting information). Photodecomposition of NOD575 was then tested in the presence of these three biological relevant reducing agents, e.g., resveratrol, cysteine, and ascorbic acid. Fluorescence turn-on was dramatically improved and completed in as short as 20 s (Fig. 5).

|
Download:
|
| Fig. 5. Spectral monitoring of the photolysis of NOD575 with 532 nm in phosphate buffer (50 mmol/L, pH 7.4) with 1% DMSO in the presence of various indicated reducing agents. | |
{kind=link}
The potentials of NOD575 for in vitro applications were tested. Pulmonary artery smooth muscle cells (PASMCs) were incubated with NOD575 (20 μmol/L) for 20 min in dark. PASMCs were then washed with phosphate buffer for 3 times. The fluorescence image before and after photolysis by green laser was acquired by a confocal microscopy. Fig. 6 indicated that cells showed no fluorescence without irradiation. After exposure to green light for 60 s, there existed strong red fluorescence from cells. These results showed that NOD575 could diffuse into PASMCs and it could release NO in vitro and the rhodamine dye generated in situ could be harnessed for imaging purposes.

|
Download:
|
| Fig. 6. Confocal fluorescence imaging of PASMCs incubated with 20 μmol/L NOD575 for 20 min. (A) Bright-field image. (B) Fluorescence image before irradiation. (C) Fluorescence image after 60 s' photo-irradiation. Scale bar: 10 mm. λex = 488 nm, λem = 555–650 nm. | |
{kind=link}
In conclusion, we have designed and synthesized a novel NO donor (NOD575) following the principle of N-nitroso push-pull dyes. It has exhibited photo-triggered release of NO upon irradiation by a green laser line at 532 nm. Also, the release of NO is accompanied by the liberation of a rhodamine fluorophore, whose fluorescence emission (λem = 575 nm) can be used as a selfcalibration mechanism for the NO release. NOD575 has a small dihedral angle of 12.67° between the N-nitroso moiety and the rhodamine scaffold by ring-closure. Such a rational design is engineered into the molecule to weaken the N-N bond and facilitate its hemolysis upon photo-irradiation. Therefore, a short exposure of ca. 100 s is sufficient to allow complete photolysis, compared to 2300s of NOD560, a similar analog with a dihedral angle of 82°. Presence of biological thiols or reducing agents does not induce the decomposition of NOD575 in dark, but can greatly accelerate its photodecomposition. Last, NOD575 is membranepermeable and compatible for in vitro biological studies.
AcknowledgmentsThe work is financially supported by the National Natural Science Foundation of China (No. 21572061) and the Fundamental Research Funds for the Central Universities (No. WY1516017).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2018.08.019.
| [1] |
P.G. Wang, M. Xian, X. Tang, et al., Chem. Rev. 102 (2002) 1091-1134. DOI:10.1021/cr000040l |
| [2] |
P.G. Wang, T.B. Cai, N. Taniguchi, Nitric Oxide Donors: For Pharmaceutical and Biological Applications, Wiley-VCH, New York, 2005.
|
| [3] |
M.R. Miller, I.L. Megson, Br. J. Pharmacol. 151 (2007) 305-321. |
| [4] |
K. Sharma, A. Iyer, K. Sengupta, H. Chakrapani, Org. Lett. 15 (2013) 2636-2639. DOI:10.1021/ol400884v |
| [5] |
H. Chakrapani, A.E. Maciag, M.L. Citro, L.K. Keefer, J.E. Saavedra, Org. Lett. 10 (2008) 5155-5158. DOI:10.1021/ol8020989 |
| [6] |
A.T. Dharmaraja, G. Ravikumar, H. Charkrapani, Org. Lett. 16 (2014) 2610-2613. DOI:10.1021/ol5010643 |
| [7] |
P.J. Shami, J.E. Saavedra, L.Y. Wang, et al., Mol. Cancer Ther. 2 (2003) 409-417. |
| [8] |
V. Calabrese, C. Mancuso, M. Calcani, et al., Nat. Rev. Neurosci. 8 (2007) 766-775. DOI:10.1038/nrn2214 |
| [9] |
M. Blangetti, A. Fraix, L. Lazzarato, et al., Chem.-Eur. J. 23 (2017) 9026-9029. DOI:10.1002/chem.v23.38 |
| [10] |
K. Kitamura, M. Kawaguchi, N. Ieda, N. Miyata, H. Nakagawa, ACS Chem. Biol. 11 (2016) 1271-1278. DOI:10.1021/acschembio.5b00962 |
| [11] |
A.C. Roveda, H. de Fazio Aguiar, K.M. Miranda, C.C. Tadini, D.W. Franco, J. Mater. Chem. B 2 (2014) 7232-7242. DOI:10.1039/C4TB00996G |
| [12] |
Z. Zhang, J. Wu, Z. Shang, et al., Anal. Chem. 88 (2016) 7274-7280. DOI:10.1021/acs.analchem.6b01603 |
| [13] |
H. He, Z. Ye, Y. Xiao, et al., Anal. Chem. 90 (2018) 2164-2169. DOI:10.1021/acs.analchem.7b04510 |
| [14] |
H. He, Y. Liu, Z. Zhou, et al., Free Radic. Biol. Med. 123 (2018) 1-7. DOI:10.1016/j.freeradbiomed.2018.04.563 |
| [15] |
P. Ning, W. Wang, M. Chen, Y. Feng, X. Meng, Chin. Chem. Lett. 28 (2017) 1943-1951. DOI:10.1016/j.cclet.2017.09.026 |
| [16] |
S. Ma, L. Li, M. She, Y. Mo, J. Li, Chin. Chem. Lett. 28 (2017) 2014-2018. DOI:10.1016/j.cclet.2017.09.027 |
| [17] |
S. Leng, Q.L. Qiao, Y. Gao, L. Miao, Z.C. Xu, Chin. Chem. Lett. 28 (2017) 1911-1915. DOI:10.1016/j.cclet.2017.03.034 |
| [18] |
X.M. Li, R.R. Zhao, Y. Yang, X.W. Lv, Y. Zhou, Chin. Chem. Lett. 28 (2017) 1258-1261. DOI:10.1016/j.cclet.2016.12.029 |
| [19] |
D. Wang, Z. Wang, Q. Zhan, et al., Engineering 3 (2017) 402-408. DOI:10.1016/J.ENG.2017.03.014 |
| [20] |
D.A. Wink, J.A. Cook, S.Y. Kim, et al., J. Biol. Chem. 272 (1997) 11147-11151. DOI:10.1074/jbc.272.17.11147 |