 2018, Vol. 29
2018, Vol. 29 



b State Key Laboratory of Precision Spectroscopy, East China Normal University, Shanghai 200062, China;
c Collaborative Innovation Center of Extreme Optics, Shanxi University, Taiyuan 030006, China
One important application of dye molecules is that they can be used as agents for photodynamic therapy (PDT) [1-3], an effective and non-invasive treatment of cancers and precancerous inductions [4-9]. These dyes used for PDT are called photosensitizers, capable of generating strongly oxidizing singlet oxygen upon photo excitation under aerobic conditions [10-13]. Therefore, a high triplet state generation yield is one key factor for dyes used in PDT. Thus, low fluorescence quantum yield and effective intersystem crossing from singlet state to triplet state are expected in photosensitizers for PDT.
PRODAN family is an important type of extrinsic fluorescence probes for pH [14], metal ions [15], biological thiols [16], oxidative species [17], and saccharides [18], etc. With the advantages of high sensitivity, specificity, fast response, and high temporal and spatial resolution, fluorescent probes is a powerful molecular tool for biological tissues imaging [19-22]. In our recent work, a new PRODAN derivative, TPZ2, was designed and synthesized [23]. This new molecule, connected together by vertical rigidity of spiro linkage, has a large two-photon absorption coefficient and it has been applied in cells, tissues and zebrafish two-photon imaging. Meanwhile, we found that TPZ2 has a fluorescence quantum yield of 0.50 in acetonitrile solution and singlet oxygen generation was observed under illumination. In order to obtain an in-depth insight into the excited state dynamics of TPZ2, we investigated the photophysics properties of TPZ2 using femtosecond transient absorption and time-correlated single photon counting (TCSPC) techniques. Unique dual excited state deactivation pathways that lead to 50% fluorescence emission and 50% triplet state generation were revealed in TPZ2.
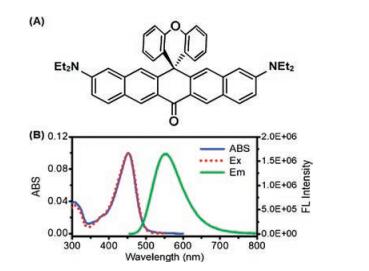
Fig. 1A shows the chemical structure of TPZ2 and the synthesis method has been described in our previous study [23]. Fig. 1B exhibits the UV–vis absorption spectra, fluorescence emission spectra and excitation spectra of molecule TPZ2 in acetonitrile solution. TPZ2 shows the absorption and emission peaks at 452 nm and 552 nm, respectively. The excitation spectra are almost identical to the absorption spectra, indicating that absorption and emission were originated from the same excited state. The fluorescence quantum yield of TPZ2 is determined to be 0.50 (Details can be found in Supporting information), suggesting it could be a good fluorescence probe for bioimaging [24].

|
Download:
|
| Fig. 1. (A) Chemical structure of TPZ2. (B) UV–vis absorption spectra (solid blue line), excitation spectra (dotted red line) and emission spectra (solid green line) of TPZ2 in acetonitrile solution. | |
{kind=link}
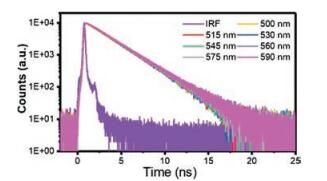
Fig. 2 shows the fluorescence decay kinetics of TPZ2 in acetonitrile solution measured by time correlated single photon counting (TCSPC) technique. The instrument response function (IRF) is determined to be ~190 ps by using Rayleigh scattering from SiO2 nanoparticle solution. The fluorescence of TPZ2 exhibits a single exponential decay with a lifetime of 2.5 ns and there is no wavelength dependence, indicating that the emission from TPZ2 is homogeneous. The excited state decay rate kEXC was calculated from the inverse of excited state lifetime τEXC from TCSPC measurement according to Eq. (1):
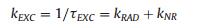
|
(1) |

|
Download:
|
| Fig. 2. Fluorescence decay curves of TPZ2 in acetonitrile solution with indicated probe wavelengths. | |
{kind=link}
Here, kRAD and kNR stand for radiative and non-radiative decay rate constants, respectively. Meanwhile, fluorescence quantum yield (QY) is defined in Eq. (2):

|
(2) |
Thus, the radiative decay rate can be calculated from the fluorescence quantum yield and the excited state decay rate kEXC (kRAD =kEXC × QY). For TPZ2, we found that both the radiative and non-radiative decay rates are 2.0 ×108 s-1.
In order to understand the excited state dynamics of TPZ2 thoroughly, femtosecond transient absorption (TA) measurements with 120 fs time resolution were performed on TPZ2. The sample was excited at 470 nm with low pump energies (0.15–0.2 μJ/pulse) in order to avoid photo-decomposition. The broadband TA spectra are shown in Fig. 3A. The TA spectra show a stimulated emission (SE) band around 560 nm, an excited-state absorption (ESA) band around 400 nm and a ground state bleach (GSB) band around 460 nm right after excitation. The SE band show a red-shift from 545 nm to 580 nm in the first 10 ps, and then this band gradually decay into a new broad and long-lived ESA band (500–750 nm) in several ns time scale. Examination of the SE band of TPZ2 shows a 0.12 eV red-shift with ~5 ps timescale (Fig. S1A in Supporting information). No iso-emissive point is seen in the SE band, indicating it is a continuous spectral shift. Best fit of the SE peak shifting dynamics reveal a time constant of 0.8 ps (Fig. S1B in Supporting information). This phenomena suggests that solvation of TPZ2 happen in the first ~1 ps and it brings TPZ2 from the FrankCondon region to the S1 minimum in the excited state energy surface. The initial ESA band centered at 400 nm also show a ~1 ps decay which should originate from the solvation dynamics and then a further ns decay is observed. The GSB signal decays very slowly but broaden a little in ns time scale. The long-lived new ESA and the non-recovered bleach signals both suggest that the initial excited state (S1) of TPZ2 could decay to a triplet state which have lifetime much longer than our instrument detection limit. Singlet oxygen generation is observed in TPZ2 (Fig. S4 in Supporting information) using the Rose Bengal as a reference, which further confirmed the existence of triplet state in TPZ2.

|
Download:
|
| Fig. 3. Transient absorption spectra (A) and kinetics (B) of sample TPZ2 in acetonitrile solution with 470 nm excitation. In the kinetics, open symbols represent the transient absorption data and the solid lines are the best fitting curves. | |
{kind=link}
Kinetic curves at representative wavelength (for ESA, GSB and SE band) were chosen to analyze the excited state dynamics of TPZ2 and the results were displayed in Fig. 3B. The ESA kinetics at 419 nm can be best-fitted by a double-exponential decay (fitting parameters in Table S1 in Supporting information) and the time constants are 1.2 ps and 2.4 ns. On the other hand, the GSB signal at 450 nm shows only one 5.1 ns time constant. At last, three time constants are needed to fit the SE band kinetics at 672 nm and the lifetimes are 1.1 ps, 2.5 ns and 5.1 ns, respectively.
As discussed above, the ~1 ps (τ1) time constants in both ESA and SE band agree with the SE peak shifting time and should be assigned to the solvation dynamics in the S1 state of TPZ2. Meanwhile, the ~2.5 ns (τ2) lifetime found in both ESA and SE kinetics matches quite well with the TCSPC data and should be assigned to the depopulation of the S1 state. At last, both GSB and SE kinetics show a 5.1 ns (τ3) time constant which correspond to the build-up of the triplet state TA signal. The intersystem crossing rate constant is calculated to be 2.0 ×108 s-1 by using equation kISC = 1/τ3. It is worth to point out that we have already determined that kNR = 2.0 ×108 s-1 for S1 state in TPZ2 from our TCSPC measurement. Since kNR is identical to kISC, intersystem crossing from S1 to T1 is the only non-radiative decay channel in TPZ2. In another word, the triplet generation quantum yield in TPZ2 is also 50%, which ensures single oxygen generation as we observed.
We also investigated the dynamics of TPZ2 in protonic ethanol solution. Fig. S2 (Supporting information) exhibits the UV–vis absorption spectra and fluorescence emission spectra of molecule TPZ2 in ethanol solution with an absorption maximum at 463 nm and an emission maximum at 581 nm, respectively. In our previous study [23], the fluorescence quantum yield of TPZ2 in ethanol solution is determined to be 0.51, almost the same to that in acetonitrile solution. The fluorescence decay measured by TCSPC also shows a single exponential decay with lifetime of 2.5 ns and it is independent of wavelength. In this case, the non-radiative decay rate kNR is also 2.0 × 108 s-1 for TPZ2 in ethanol. The transient absorption spectra of TPZ2 in ethanol solution were also measured and the spectra profile are similar to those seen in acetonitrile solution (Fig. S3B). The ISC kinetics of TPZ2 in acetonitrile and ethanol solution at 672 nm shows a similar decay (Fig. S3C in Supporting information), and the time constants of ISC (τ3) is found to be 5.3 ns (Table S1). Thus, the ISC rate constant of TPZ2 in ethanol is 1.9 × 108 s-1. Again, the non-radiative rate is in agreement with the ISC rate under experimental uncertainty, suggesting that the ISC is also the only non-radiative decay channels in TPZ2 in ethanol.
Bioimaging needs high fluorescence quantum yield [24], while photodynamic therapy wants low fluorescence quantum yield and high singlet oxygen yield [25]. It is well-known to us that the two pathways of fluorescence emission and triplet state generation are two opposite process, thus, a sample with both high fluorescence quantum yield and high triplet state yield is unusual. Here, the ratio between these two yields in TPZ2 is 1:1, which means that this molecule is an ideal fluorescence probe for bioimaging as well as a potential photodynamic therapy agent.
According to our TCSPC and TA data, we proposed an excited state relaxation mechanism for TPZ2 as displayed in Fig. 4. The purple arrow represents 452 nm excitation. After initial excitation, TPZ2 decay from Frank-Condon region to the minimum of S1 through solvation and the lifetime is ~1 ps. While the S1 state of TPZ2 show a 2.5 ns lifetime, the intersystem crossing lifetime is 5.1 ns. This result indicates that there are dual excited state decay pathways for S1 state of TPZ2: One is emissive pathway that involves fluorescence from S1 to the ground state and the other is non-emissive pathway which involves intersystem crossing from S1 to T1. Thus, the radiative and non-radiative decay rate constants of the S1 state in TPZ2 are equal and number is determined to be 2.0 × 108 s-1. Triplet state of TPZ2 can be quenched by oxygen in the solution, leading singlet oxygen generation.

|
Download:
|
| Fig. 4. Proposed excited state deactivation mechanism of TPZ2 in solution. | |
{kind=link}
In conclusion, excited state dynamics of TPZ2 has been investigated by TCSPC and femtosecond transient absorption. Unique dual excited state deactivation pathways were discovered in TPZ2. Half of the excited state energy will dissipate as fluorescence and the lifetime is found to be 2.5 ns. The other half energy can transfer into triplet state through intersystem crossing and further generate singlet oxygen in solution. The special excited state relaxation mechanism ensures that TPZ2 could have both high fluorescence quantum yield and singlet oxygen generation yield, providing us a powerful dye for potential application in bioimaging and photodynamic therapy in the same time.
AcknowledgmentThis study was funded by the National Natural Science Foundation of China (No. 11674101).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j. cclet.2018.08.008.
| [1] |
E. Van de Wincke, M. Mascaraque, A. Zamarron, et al., Adv. Funct. Mater. 28 (2018) 1705938. DOI:10.1002/adfm.v28.24 |
| [2] |
L. Liu, T.W. Li, Z. Ruan, L.F. Yan, J. Mater. Sci. 53 (2018) 9368-9381. DOI:10.1007/s10853-018-2276-6 |
| [3] |
S.S. Kelkar, T.M. Reineke, Bioconjugate Chem. 22 (2011) 1879-1903. DOI:10.1021/bc200151q |
| [4] |
Y.Z. Shen, A.J. Shuhendler, D.J. Ye, J.J. Xu, H.Y. Chen, Chem. Soc. Rev. 45 (2016) 6725-6741. DOI:10.1039/C6CS00442C |
| [5] |
C. Seidl, J. Ungelenk, E. Zittel, et al., ACS Nano 10 (2016) 3149-3157. DOI:10.1021/acsnano.5b03060 |
| [6] |
E. Ju, K. Dong, Z. Chen, et al., Angew. Chem. Int. Ed. 55 (2016) 11467-11471. DOI:10.1002/anie.201605509 |
| [7] |
S. Julie, H. Valérie, S. Angélique, et al., Angew. Chem. Int. Ed. 54 (2015) 169-173. DOI:10.1002/anie.201407537 |
| [8] |
T.J. Dougherty, J. Clin. Laser Med. Surg. 20 (2002) 3-7. DOI:10.1089/104454702753474931 |
| [9] |
M.A. Maccormack, Semin. Cutan. Med. Surg. 27 (2008) 52-62. DOI:10.1016/j.sder.2007.12.001 |
| [10] |
C.W. Brian, S.P. Michael, Phys. Med. Biol. 53 (2008) R61. DOI:10.1088/0031-9155/53/9/R01 |
| [11] |
T.J. Dougherty, C.J. Gomer, B.W. Henderson, et al., Natl. Cancer Inst. 90 (1998) 889-905. DOI:10.1093/jnci/90.12.889 |
| [12] |
C. Schweitzer, R. Schmidt, Chem. Rev. 103 (2003) 1685-1758. DOI:10.1021/cr010371d |
| [13] |
W. Fan, P. Huang, X. Chen, Chem. Soc. Rev. 45 (2016) 6488-6519. DOI:10.1039/C6CS00616G |
| [14] |
P.H. Jung, L.C. Su, K.E. Sun, et al., Angew. Chem. Int. Ed. 51 (2012) 2673-2676. DOI:10.1002/anie.201109052 |
| [15] |
K.H. Myung, C.B. Rae, Chem.-Asian J. 6 (2011) 58-69. DOI:10.1002/asia.201000542 |
| [16] |
S. Singha, D. Kim, A.S. Rao, et al., Dye. Pigment. 99 (2013) 308-315. DOI:10.1016/j.dyepig.2013.05.008 |
| [17] |
X. Dong, C.H. Heo, S. Chen, et al., Anal. Chem. 86 (2014) 308-311. DOI:10.1021/ac403226h |
| [18] |
T.Y. Shun, L.H. Yeon, L.C. Su, et al., Angew. Chem. Int. Ed. 48 (2009) 8027-8031. DOI:10.1002/anie.v48:43 |
| [19] |
H. Zhu, J. Fan, J. Du, X. Peng, Acc. Chem. Res. 49 (2016) 2115-2126. DOI:10.1021/acs.accounts.6b00292 |
| [20] |
X. Jiao, Y. Li, J. Niu, et al., Anal. Chem. 90 (2017) 533-555. |
| [21] |
B. Zhu, L. Wu, Y. Wang, et al., Sensor. Actuat. B-Chem. 259 (2018) 797-802. DOI:10.1016/j.snb.2017.12.135 |
| [22] |
W. Sun, S. Guo, C. Hu, et al., Chem. Rev. 116 (2016) 7768-7817. DOI:10.1021/acs.chemrev.6b00001 |
| [23] |
Z. Lei, P. Yue, X. Wang, et al., Chem. Commun. 53 (2017) 10938-10941. DOI:10.1039/C7CC06031A |
| [24] |
Y. Yang, Q. Zhao, W. Feng, F. Li, Chem. Rev. 113 (2013) 192-270. DOI:10.1021/cr2004103 |
| [25] |
A. Kamkaew, S.H. Lim, H.B. Lee, et al., Chem. Soc. Rev. 42 (2013) 77-88. DOI:10.1039/C2CS35216H |