 2018, Vol. 29
2018, Vol. 29 



Functional materials have become very important in the study of life processes. Over the past decade, a wide variety of novel chromic materials comprised of organic fluorescent compounds have been developed and applied in the construction of sensors [1-5], safety devices [6], and imaging [7-12] and optoelectronic systems [13, 14]. Most of these luminescent materials display versatile optical responses to different external stimuli with color changes from one to another or between emerging and disappearing, and as a result, they are useful for translating macroscopic natural phenomena like mechanical force, heat, light, electricity and chemical processes into optical changes [15-19]. For the most part, these materials have been designed to emit fluorescence or phosphorescence in the UV or visible regions so that it can be observed using readily available instrumentation or the naked eye. In contrast, very few studies have described photoluminescent materials that emit long wavelength light in the near-infrared (NIR) region despite the fact that materials of this type would have potential applications in systems for highly sensitive and optical bioimaging and biomolecule detection [20-25]. Notably the NIR region here is referring to NIR-Ⅰ which defines from 740 nm to 1000 nm, and the following samples displayed are selected for their emission bands covering the NIR region, not only with the maximum emission peak locating at the region.
Aggregation induced emission (AIE), described originally by Tang, is a phenomenon in which planar organic compounds display highly efficient light emission in their crystalline and aggregated states [26]. The advent of AIE has opened the new door to the discovery of new solid fluorescent materials.
Luminescent properties are very sensitive to subtle changes that take place in materials. In most cases, responses of solids to stimuli cause bathochromic shifts in emission maxima as a consequence of changes occurring in molecular packing assemblies or chemical structures. Several strategies have been utilized to design materials that undergo emission wavelength changes that extend into the NIR region [27-29]. However, though the methods are relatively mature, the challenge still take place in the unpredictable optical changes to long wavelength of all various structures, which means lacking of a universal method. In this review, we describe selected systems of this type, concentrating for the most part on those in which emission changes are promoted by mechanical stimuli.
2. MechanochromismMechanochromism is the phenomenon associated with changes in absorption and/or fluorescent colors that occur in response to external mechanical forces such as grinding, pressing and stretching. The photophysical properties of materials that undergo mechanochromism in the solid state rely heavily on their molecular structures as well as on the modes of assembly and packing, which impact hydrogen bonding, CH-π and metal-metal interactions. In most cases, mechanical stimuli bring about changes in packing of molecules in the crystalline phase of solids. Hence it is interesting to explore the inherent correlation that might exist between stimuli and emissive properties.
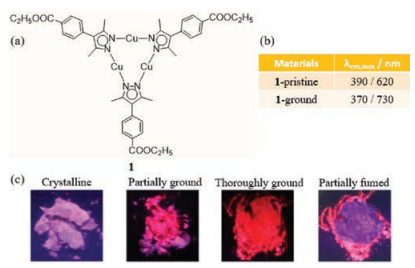
In 2014, Li et al. demonstrated that well-known copper pyrazole complexes display deep red emission bands (λem, max > 700 nm) upon mechanical stimulation [30]. Upon being subjected to grinding, the complex Cu3(EBPz)3 (1 where EBPz is ethyl-4'-benzoate-3, 5-dimethylpyrazolate, Fig. 1a) undergoes an emission color change from blue-violet to red. Specifically, the color change is associated with a shift in two emission maxima from 390 nm (high energy (HE) band) and 620 nm (low energy (LE) band) for pristine 1 to 370 nm and 730 nm for ground 1 (Fig. 1b). While this change is reversed by addition of ethanol (Fig. 1c). The presence of the para-ester auxochrome is responsible for the special dual emission property of 1 in the solid state. The emission color changes displayed by this complex are caused by a mechanism that is different from those promoted by alterations in packing modes, it roots in the energy transfers between energy bands. The LE band corresponding to phosphorescence and the HE band associated with fluorescence appear simultaneously. Moreover, the relative intensities of these emission bands in the crystalline sample is HE > LE while in the ground amorphous solid the relative intensities are LE > HE, and the LE lifetime is longer. In accord with previous reports, the change in the energies and relative intensities of the emission bands in 1 brought about by grinding can be attributed to shortening of the intermolecular Cu-Cu distances that results in enhancement of phosphorescence from the metal-metal triplet states.

|
Download:
|
| Fig. 1. (a) The chemical structures of 1. (b) Solid-state optical properties of 1 before and after grinding. (c) Photographs of samples of 1. Reprinted with permission [30]. Copyright 2014, American Chemical Society. | |
{kind=link}
In 2017, Ito et al. reported the discovery of the gold(Ⅰ) isocyanide complex 2 (Fig. 2a) which exhibits a surprising bathochromic shift of its emission maximum upon mechanical stimulation [31]. Two different crystalline forms of this substance, 2a and 2b, exhibit respective mechanofluorochromic changes from 448 nm to 900 nm and 710 nm to 900 nm upon being transformed from their pristine to ground states with the fluorescence color turning dark (Fig. 2b). The strategy employed to design these complexes was developed in preliminary studies, which showed that extending conjugation in the π-segment of the aryl moiety causes a red-shift of the emission band. The results of crystal analysis showed that the two polymorphs have similar packing modes even though the differences exist between their emission properties. Specifically, the dihedral angle between the anthracene plane and the phenyl ring in 2a is 86.17° while the corresponding angle in 2b is 50.84°. Thus, in the crystalline state of 2a emission arises from a π-π* transition that is localized on the anthracene moiety. Moreover, the smaller dihedral angle in 2b enables phosphorescence to occur from a ligand-to-ligand charge transfer (LLCT) state generated by intramolecular charge transfer from the anthracene to the aryl isocyanide moiety. Though 2a exhibits an unexpected short wavelength emission maximum at 448 nm, the magnitude of the shift promoted by grinding (Δλem, max = 452 nm) is unprecedented. Powder X-ray diffraction (PXRD) patterns of the pristine samples contain sharp diffraction peaks associated with crystalline states, which disappear upon grinding, indicating that both 2a and 2b undergo crystalline-to-amorphous phase transitions induced by mechanical stimulation. Compared with another polymorph 2c, which only displays a broad emission maximum at 889 nm, the shifts into the NIR region brought about by grinding 2a and 2b are caused related to strengthen in intermolecular interactions such as aurophilic interactions (Fig. 2c), which can also be seen by using IR absorption spectroscopy.

|
Download:
|
| Fig. 2. (a) The chemical structure of 2. (b) Solid-state optical properties of 2a and 2b before and after grinding. (c) Schematic representation of the emission mechanism in 2a/2b and 2ground. Reprinted with permission [31]. Copyright 2017, American Chemical Society. | |
{kind=link}
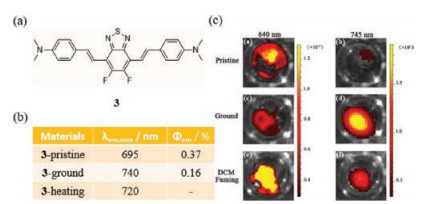
In addition to the metal complexes described above, it seems there are more organic compounds in the reported cases. Our group recently studied the difluorobenzothiadiazole-based fluorescent material DFBTN (3, Fig. 3a) [32]. This substance, which has a donor-π-acceptor-π-donor (D-π-A-π-D) structure, displays reversible mechanofluorochromic behavior in the solid state from red to dark. The fluorescence maximum of 3 shifts from 695 nm to 740 nm in the NIR region upon mechanical stimulation promoted by grinding (Fig. 3b). It is worth noting that this change can be reversed by fuming the ground sample with CH2Cl2 as well as partially by thermal annealing (Fig. 3c). Analysis of its crystal structure shows that 3 is highly planar, a property that facilitates intramolecular charge transfer from the electron rich p-aminobenzene groups to the benzothiadiazole core. The results of PXRD analysis suggest that a crystalline-to-amorphous phase transition occurs upon grinding and that the original crystalline state is regained by exposure to CH2Cl2 vapor. Furthermore, the results of TD-DFT calculations show that the occurrence of molecular sliding in dimeric subunits is the cause of the bathochromic-shift in emission promoted by grinding.

|
Download:
|
| Fig. 3. (a) The chemical structure of 3. (b) Solid-state optical properties of 3 before and after grinding. (c) Photographs of a Xenogen IVIS spectrum images of 3 upon asprepared (a, b), grinding (c, d) and CH2Cl2 fuming (e, f); (a, c, e) λex = 470 nm, λem = 640 nm; (b, d, f) λex= 470 nm, λem = 745 nm. Reprinted with permission [32]. Copyright 2018, WILEY-VCH. | |
{kind=link}
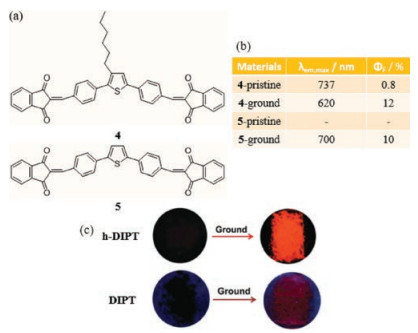
Besides compound 3, the opposite structure referring to acceptor-π-donor-π-acceptor (A-π-D-π-A) appeared to show similar chromic properties in the NIR region. Two novel A-π-D-π-A substances, 4 and 5, which contain a diphenylthiophene core and two 1, 3-indandione side chains linked by double bonds, were investigated by Zhao et al. (Fig. 4a) [33]. A-π-D-π-A luminophores reported previously were found to have poor mechanochromic properties owing to their highly twisted and rigid conformations in aggregated states. In contrast, DFT analysis shows that 4 and 5 adopt slightly twisted conformations, which enable them to display mechanochromic responses. The authors applied one approach to fabricate 1D rods of these substances involving recrystallization (S-process) and another to create 0D particles through rotary evaporation (Q-process). Experiments with these materials reveal that crystals of h-DIPT (4), formed using the Sprocess, display shorter wavelength emission at 640 nm, while longer wavelength (737 nm) and weaker emission is exhibited by crystals fabricated using the Q-process. These phenomena demonstrate that differences in micro morphologies, brought about by differences in the tightness of molecular arrangements through alterations in the speed of crystal formation, can lead to differences in emission spectral characteristics. The 0D particles of 4 exhibit an interesting mechanochromic phenomenon from dark to light red corresponding to blue shifting of emission back to 620 nm. In addition, in contrast to unground particles that are nonemissive, ground 5 displays a new intense peak at 700 nm in NIR region (Figs. 4b and c). The results of conformational investigations show that, because of the different speeds of assembly, the Q-process results in generation of a metastable crystalline phase with J-aggregation. This contrasts with the highly crystalline herringbone packing seen in the 1D form of 5, where a more planar conformation exists and leads to emission quenching. Upon mechanical stimulation, the original states of these substances are transformed into amorphous solid states that are strongly emissive. The results of this effort, suggest that strategies for constructing slightly twisted conformations in the solid state might offer new avenues to design NIR fluorochromic materials.

|
Download:
|
| Fig. 4. (a) The chemical structure of 4 and 5. (b) Solid-state optical properties of 4/5 before and after grinding. The samples were both 0D particles. (c) Photographs of samples of 4 and 5. Reprinted with permission [33]. Copyright 2016, Royal Society of Chemistry. | |
{kind=link}
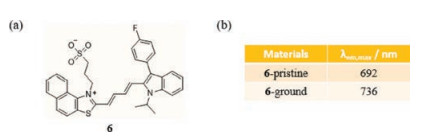
Not only the type of conjugated structures makes sense, but other luminogens have been employed to construct long wavelength emitting materials. In 2017, Yang et al. demonstrated that the naphthothiazolium framework can be used to create NTPS-FPI (6) (Fig. 5a), which in the solid state emits light with a maximum emission wavelength at 692 nm [34]. This compound displays mechanofluorochromism from deep-red to dark in the form of a red-shift of emission to 736 nm promoted by grinding (Fig. 5b). Similarly, 6 undergoes a phase transition that is attributed to destruction of crystallinity and the creation of amorphism. Additionally, a reversible transition occurs through organic vapor fuming of the ground material, and the entire cycle of grindingfuming can be carried out for more than three times. To further investigate its microstructures and packing modes, 6 was subjected to single crystal X-ray diffraction analysis. The results show that, surprisingly, besides C-H…O and C-H…F hydrogen bonds, an extra solvent molecule CH3OH is present and through C-H…O bonds it facilitates stabilization of the packing structure. These multiple interactions hinder rotation of the molecules in the crystal, thus causing intense emission in the aggregated states. Furthermore, the results of other measurements confirm that a transition from a thermodynamically stable, highly organized phase to a metastable state occurs upon mechanical stimulation of pristine 6.

|
Download:
|
| Fig. 5. (a) The chemical structure of 6. (b) Solid-state optical properties of 6 before and after grinding. | |
{kind=link}
The emission bands of other kinds of materials also undergo different shifts in conjunction with emission in the NIR region when subjected to mechanical stimuli. For example, there are some reports exhibiting intensity changes in the NIR region which result in chromic phenomena. Early in 2014, Wang et al. described a group of organoboron compounds, 7-10 (Fig. 6a), that display fluorescent "ON-OFF" responses in the NIR region from bright to dark [35]. For example, 7 crystallizes from solution as solid having a strongly red-shift emission maximum at 728 nm (Fig. 6b). This was the first example of crystalline-enhanced emission (CEE) of this kind of structures in the NIR region. The observations show that mechanical grinding transforms an ordered to a random array of these powders in association with the vanishing of fluorescence (Fig. 6c). Importantly, 7-10 all show reversible mechanofluorochromic responses that can be reversed by CH2Cl2 fuming, where the ground solids recover their original crystalline form along with recovering their strong emission. Surprisingly, a spin-coated thin film of 7 is nearly non-emissive. Because both the powder state and film contain 7 in its solid state, the huge optical difference between the two situations is ascribed to differences in morphologies. Accordingly, the powder contains a crystalline state the displays sharp diffraction peaks whereas the film is comprised of amorphously packed 7. In addition, acid/base vapors promote fluorescence changes. Owing to the presence of a dimethylamine group in each, simple treatment of crystalline 7-10 trifluoroacetic acid (TFA) fumes induces fluorescent quenching. Moreover, the formed non-emissive solid is transformed to a NIR emitting analogue by treatment with triethylamine (TEA) vapor (Fig. 6c). These findings are a consequence of blocking and unblocking intramolecular charge transfer by adjusting the protonation state of the amine moiety.

|
Download:
|
| Fig. 6. (a) The chemical structure of 7-10. (b) Solid-state emissive properties of 7-10 and changes after grinding and fuming. (c) PL spectra of ground/annealed samples (left) and TFA/TEA fumed samples (right) 7. Inside: photographs of repeated switching between dark and bright solid states. Reprinted with permission [35]. Copyright 2014, American Chemical Society. | |
{kind=link}
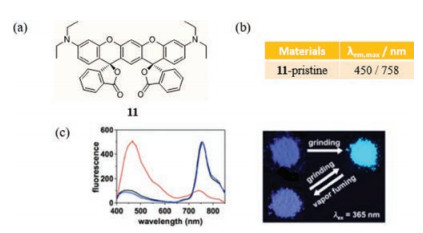
In 2015, Enomoto et al. described a new type of mechanochromic organic substance aminobenzopyranoxanthene (11, cis-ABPX01°), which contains a spirolactone moiety (Fig. 7a) [36]. This substance displays a dual fluorescence behavior. Specifically, the emission spectrum of the pristine solid contains a peak centered at around 450 nm and a NIR band with an emission maximum at 758nm, corresponding to a large Stokes shift of over 400 nm (Fig. 7b). It is noteworthy to mention that the long wavelength emission at 758nm is only characteristic of the solid form and does not exist even in spectra of highly concentrated solutions of 11 that emit blue fluorescence. Analysis shows that molecules of 11 in the NIR emitting crystalline form exist in an antiparallel slip-stacked dimer mode with additional solvent molecules. In contrast, blue fluorescence is believed to stem from the monomer configuration. After eliminating the possibility that triplet excitons, like those involved in the known analogue rhodamine, play a role in this process, the authors conclude that NIR emission in the solid is associated with photoinduced opening of the spiro-ring system. Moreover, the subtle structural change taking place in 11 favors formation of a planar slip packing. As for mechanical stimuli responses, the NIR emission reduces heavily while the weak short-wavelength emission is strongly strengthened with the fluorescence color altering from deep purple to sky blue (Fig. 7c). However, an additional finding reveals that the ground powder of 11 has optical properties that are similar to those of a solvent-desorbed sample. It means that the treatment with CH2Cl2 causes a solvent molecule to be regained in the ground solid and recovery of the original fluorescence emission.

|
Download:
|
| Fig. 7. (a) The chemical structure of 11. (b) Solid-state emissive properties of 11 including CH2Cl2 molecules. (c) PL spectra of ground (red)/fumed (blue) samples (left) and photographs samples of switching between deep purple and sky blue (right). Reprinted with permission [36]. Copyright 2015, American Chemical Society. | |
{kind=link}
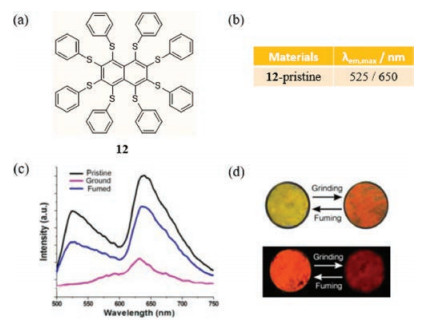
In 2017, Zhu et al. investigated the optical properties of the phenylthio-substituted naphthalene derivative 12 (Fig. 8a), which was designed to contain adjustable C-H…π interactions for selfassembly [37]. Naphthalene derivative 12 has both an intramolecular charge transfer (ICT) and a planar local excited state, which result in a dual emission including a HE fluorescence band at 525 nm and a LE phosphorescence band at 650 nm in the NIR region (Fig. 8b). Moreover, it was anticipated that C-H…π interactions of the face-to-face assembly patterns in the solid state would differ depending on its aggregation states and that a tight aggregation state would display a stronger NIR phosphorescence band. Surprisingly, the opposite response is observed. Specifically, grinding crystals of 12 causes the intensity of the phosphorescence NIR bands to decrease while the fluorescence band nearly completely vanishes, so the luminescence turns from orange to red. Upon fuming in ethanol vapor, emissions of the pristine crystals of 12 reappear but with weaker intensities (Figs. 8c and d).

|
Download:
|
| Fig. 8. (a) The chemical structure of 12. (b) Film-state emissive properties of 12 made with ethanol. (c) PL spectra of films made with ethanol. (d) Photographs samples of switching. Reprinted with permission [37]. Copyright 2017, American Chemical Society. | |
{kind=link}
In the same time, although rare, some hypsochromic responsive materials that originally display NIR emissions also exist. In 2017, Yang et al. described the 1, 4-diketo-pyrrolo[3, 4-c]-pyrrole derivative (CODPP) 13, which contains a terminal carbazole donor group linked by an oxadiazole (Fig. 9a) [38]. Crystals of 13 undergo grinding-induced mechanofluorochromism involving a change from a state that emits NIR light with a maximum at 723 nm to one that has a red fluorescence maximum at 614 nm. Unlike the cases described above, the original emitting property cannot be regenerated by either fuming or heat annealing, but both the pristine and ground crystals undergo a heat-induced change to form a state that displays a yellow fluorescence at 556nm (Figs. 9b and c). The results of calculations demonstrate that strong distortions exist among aromatic rings of three building blocks in 13, which restrain intramolecular charge transfer. Therefore, the special fluorochromic phenomenon observed for this material can be attributed to subtle changes of the degree of π-π overlap of molecules in the crystals. The authors suggest that a more planar core and greater π-π interactions between face-to-face parallel packed molecules exist in the NIR emitting state of 13, while twist conformations with lowered π-π interactions exist in the yellow emitting state. Although not proven using crystal analysis techniques, it is suggested that both states generate an amorphous red state upon mechanical stimulation, while return to a thermodynamically stable yellow fluorescence state occurs upon heat annealing.

|
Download:
|
| Fig. 9. (a) The chemical structure of 13. (b) Solid-state emissive properties of 13 before and after grinding, heating. (c) Photographs samples of switching. Reprinted with permission [38]. Copyright 2017, Royal Society of Chemistry. | |
{kind=link}
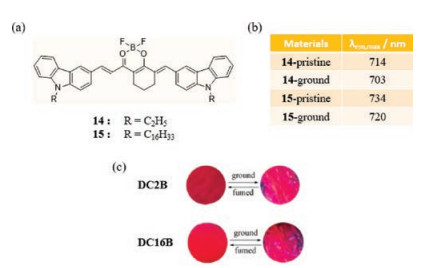
Lu et al. recently developed two difluoroboron β-diketonate derivatives, DC2B (14) and DC16B (15), in which carbazole units are linked by vinyl groups (Fig. 10a) [39]. The results show that 14 emits red NIR light with a maximum centered at 714 nm. Upon grinding crystals of this substance, the emission band shifts to 703 nm and the fluorescence turns rose-red. Likewise, the emission maximum of 15 changes from 734 nm to 720 nm upon grinding (Fig. 10b). The emission changing process for both substances can be reversed by fuming or heat annealing (Fig. 10c). There is an extra element participating in the force perturbation through grinding. It is found the isolated and aggregated units are both involved in initial pristine samples of 14, and in ground solid the ratio of two units changes and naturally impacts the fluorescent colors.

|
Download:
|
| Fig. 10. (a) The chemical structures of 14 and 15. (b) Solid-state emissive properties of 14/15 before and after grinding. (c) Photographs samples of switching. Reprinted with permission [39]. Copyright 2018, Elsevier. | |
{kind=link}
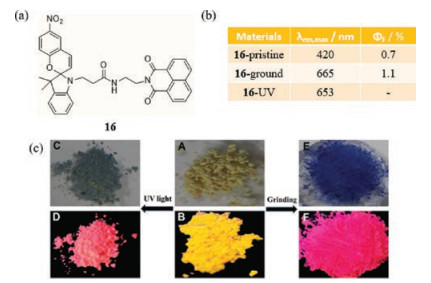
In each of the systems described above, mechanical stimulation causes a crystalline-to-amorphous transition. However, Yin et al. developed the dyad 16 (Fig. 11a), which undergoes mechanofluorochromism from yellow to reddish associated with formation of a NIR emission band that is caused by a crystalline to crystalline transformation [40]. The crystalline state of this substance, which contains the spiropyran (SP) group in its ring-closed form, displays an emission band centered at 420 nm. Quantized grinding of 16 at 32 MPa causes the development of a new fluorescence band located at 665 nm and extending to about 750 nm, which is ascribed to the SP ring-open form. Heating causes regeneration of the initial fluorescence band. In addition, UV irradiation of the 420 nm emitting crystals of 16 also leads to a red shift in emission to 653 nm (Figs. 11b and c). Observations made in a number of experiments suggest that morphological changes are responsible for the optical changes. Specifically, the as-prepared sample of 16 is comprised of clear self-assembled fibrillary aggregates, which change to disordered spherical structures upon grinding. XRD data suggest that the transition is crystalline to crystalline, because the sharp peaks in the XRD spectrum remain following grinding. However, the mode of assembly of 16 does not change upon UV irradiation that promotes SP ring opening. The forces causing the morphological change arise from hydrogen bonding and intermolecular π-π interactions between the naphthalimide cores after grinding.

|
Download:
|
| Fig. 11. (a) The chemical structure of 16. (b) Solid-state emissive properties of 16 before and after grinding, and under UV irradiation. (c) Photographs samples of switching. (A, ·B) the original powder. (C, ·D) powder after UV irradiation. (E, ·F) powder after grinding under natural light and 365 nm UV light respectively. Reprinted with permission [40]. Copyright 2017, WILEY-VCH. | |
{kind=link}
Current studies are underway to broaden the range of mechanofluorochromic materials [41]. Though the general mechanisms responsible for the emission changes that occur are believed to involve morphological and packing alterations, the inherent relationships between these changes and molecular structures are less clear.
3. PhotochromismPhotofluorochromic materials are compounds that possess the ability to undergo emission color changing photoisomerization upon exposure to external light. Emission changes of most of these materials are cause by photocyclization reactions that are promoted by irradiation with ultraviolet light and reversed by irradiation with visible light. The conformational changes caused by the bond forming and cleaving reactions are the source of the changes in absorption and emission wavelengths and/or intensities. Additionally, in terms of practical applications, these types of compounds display extraordinary fatigue resistance and a high sensitivity to light stimuli, combining to make them typical turnon functional materials.
In 2013, Kim et al. reported the results of a study of integrated binary nanococktails (NCs) comprised of the photochromic bisthienylethene (BTE) derivative 17 and the cvCP polymer 18 containing the DOP-NC moiety 19 (Fig. 12a) [42]. In the solid state, the cvCP group displays an intense NIR fluorescence centered at about 728 nm owing to AIE. Moreover, the BTE derivative exhibits photoswitching between a UV-absorbing colorless ring-open form and a ring-closed isomer that has broad absorption into the NIR region. Photoswitchable nanoprobes are formed by mixing these two compounds. The author succeeds in developing the new photochromic probe, DOP-NC, which is well-suited for imaging in vivo (Fig. 12b). The physically self-assembled nanoconstruct displays bright fluorescence in the NIR region when exposed to visible light at 655 nm. This composite becomes nonfluorescent upon UV irradiation at 254 nm because of the operation of efficient intraparticle fluorescence resonance energy transfer (FRET) (Fig. 12c). The probe was employed to produce repeated imaging signals in a mouse model.

|
Download:
|
| Fig. 12. (a) The chemical structures of 17, 18 and 19. (b) Schematic representation of the cvCP/BTE integrated binary NCs and their photoswitching reaction by UV/visible light illumination. (c) Reversible photoswitching modulation of DOP-NC embedded in the resected lymph node. Reprinted with permission [42]. Copyright 2013, WILEY-VCH. | |
{kind=link}
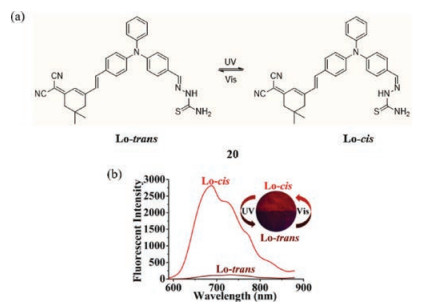
Zhou et al. developed the thiosemicarbazide Schiff base Lo (20) (Fig. 13a), which displays photochromic behavior promoted by an E-Z isomerization about the C=N bond in the solid state stimulated by UV-vis light [43]. Exposure of 20 to 365 nm irradiation generates the cis isomer that exhibits strong emission extending into the NIR region showing red fluorescence (> 700 nm). However, switching to the trans isomer results in much less intense emission with bands centered at 650 nm and 800 nm (Fig. 13b). TD-DFT calculations are performed to gain more insight into the photophysical and photochemical properties of this substance.

|
Download:
|
| Fig. 13. (a) The chemical structure of 20. (b) PL spectra of isomeric samples. Inside: photographs samples of isomeric switching under UV irradiation. Reprinted with permission [43]. Copyright 2016, Royal Society of Chemistry. | |
{kind=link}
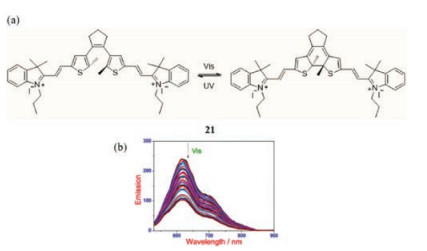
We later observed that the dithienylethene derivative 21, containing A-π-A moieties (Fig. 14a), in solution displays decreasing emission intensities in the range of 550-850 nm upon irradiation using visiblelight(Fig. 14b), accompanied with a darker fluorescence[44]. In contrast to other analogous systems, visible rather than UV light promotes photocyclization of 21 togenerate the ring-closed isomer. The molecule could also exclusively responds to the cyanide anion basing on the forming of indole-based dithienylethene.

|
Download:
|
| Fig. 14. (a) The chemical structure of 21. (b) PL spectra of samples in solution under light irradiation at 402 nm. Reprinted with permission [44]. Copyright 2015, American Chemical Society. | |
{kind=link}
As it can been seen from the compounds discussed above, the photochromic NIR materialsstill rooted mainly inthe traditional on-off structures. On account of the usual intensity changes in this type of chromic substances, the key point is still to construct pristine emissive NIR structure, so it is easier to achieve the aim of subsequent photoswitching through bond forming or cleaving, isomerism, FRET and so on. Since the method of ring-opening or closing is binding tight to specific structures, isomerism relies much on the material itself, the way utilizing FRET while combining different functional parts seems to be more flexible and practical. Moreover, it should be stressed that subtle structural changes are able to lead huge leaps.
4. ThermochromismThermofluorochromism is the phenomenon involving thermal generation of color changes associated usually with a change in the state of aggregation of a substance and its effect on interactions between the chromophoric units in solution. Only a few materials that display thermofluorochromism in the solid state have been uncovered to date.
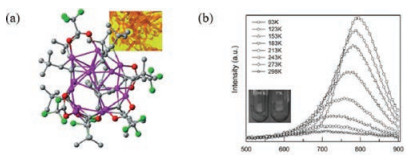
In 2016, Zheng reported that the high-nuclear CuI-alkynyl cluster [Cu15I(tBuC≡C)10(CF3COO)5]·tBuC≡CH (22), having a pentagonal bipyramid structure, displays thermochromism in association with formation of a NIR emission band in solid state [45]. This coordination complex undergoes a large shift in its fluorescence emission maximum from 710 nm at 298 K to 793 nm at 93 K, along with a large 17-fold increase in intensity, while the strong NIR fluorescence appear (Fig. 15). By using variable-temperature XRD experiments, it is shown that the Cu-Cu distance decreases and, as a result, overlap between the s and p atomic orbitals increases as the temperatureis decreased.This causes loweringof the energyfor the cluster-centered (3CC) transition. This process is typically observed in other CuI and AuI clusters.

|
Download:
|
| Fig. 15. (a) The ball-and-stick representation of cluster 22. Inside: optical microscope picture of crystals of 22. (b) PL spectra of samples under different temperature conditions. Reprinted with permission [45]. Copyright 2016, WILEY-VCH. | |
{kind=link}
5. Other chromism
Other types of chromic fluorescence behaviors have been observed, including vapochromism, halochromism and aggregachromism. In the examples 7-10 described above, we describe fluorescence changes that are a consequence of treatment with acid/base vapors. In another example of this phenomenon, Würthner et al. developed perylene bisimides derivatives that show decreasing NIR fluorescence in the presence of tetrabutylammonium hydroxide (TBAH) in solutions, which can be recovered by addition of TFA [46]. Moreover, Wang's and Zhao's groups independently reported halochromic NIR materials that are stimulated by addition of Bronstead acids [47, 48]. Furthermore, Chen et al. developed a series of metal complexes that respond to volatile organic compounds by undergoing changes in their fluorescence characteristics [49]. Yang et al. described a pair of combined D-A butterflies that display aggregation enhanced emission (AEE) around 775 nm in the solid state, which is blueshifted to 644 nm by heat annealing [50]. Meanwhile, grinding samples of these materials after annealing causes a red shift of their fluorescence maxima back to that of pristine materials.
6. ConclusionsAs described above, several chromic luminescent materials that emit light in the NIR region in response to various external stimuli have been developed. Because of the scarcity of examples, this article focus mainly on mechanochromism, which involves changes in fluorescence and phosphorescent properties brought about by mechanical forces. We have attempted to shed light on the mechanism responsible for the emission changes by providing several concrete examples.
Thus far, several approaches have been utilized to generate new NIR emitting materials, including increasing the π-conjugation area, enhancing charge transfer effects, introducing the possibility for twisted-planar conformation changes. By considering the systems studied to date, it is possible to conclude that phase transitions associated with changes in packing modes and molecular interactions is the most common mechanism for mechanochromism. However, conformation changes, morphological alterations and even changes in protonation states also promote emission changes.
Although at this point it is not possible to propose a universal picture that describes chromism, practical applications of materials that display this propertyare beginning to appear. There is little doubt that responsive NIR materials will become valuable tools in imaging, medical treatment, security devices and responsive sensors.
AcknowledgmentsWe acknowledge the National Natural Science Foundation of China (Nos. 21676113, 21402057, 21772054, 21472059), Distinguished Young Scholar of Hubei Province (No. 2018CFA079), Youth Chen-Guang Project of Wuhan (No. 2016070204010098) for the financial support. This work is also supported by the 111 Project (No. B17019), the Ministry-Province Jointly Constructed Base for State Key Lab-Shenzhen Key Laboratory of Chemical Biology (Shenzhen), the State Key Laboratory of Materials-Oriented Chemical Engineering (No. KL17-10), Open Project Fund of Key Laboratory of Natural Resources of Changbai Mountain & Functional Molecules, Yanbian University (No. NRFM201701), Ministry of Education, the foundation of Key Laboratory of Synthetic and Biological Colloids, Ministry of Education, Jiangnan University (No. JDSJ2017-07), self-determined research funds of CCNU from the colleges' basic research and operation of MOE (No. CCNU18TS012).
| [1] |
Y. Shiraishi, R. Miyamoto, T. Hirai, Org. Lett. 11 (2009) 1571-1574. DOI:10.1021/ol900188m |
| [2] |
E.P. Chan, J.J. Walish, E.L. Thomas, C.M. Stafford, Adv. Mater. 23 (2011) 4702-4706. DOI:10.1002/adma.201102662 |
| [3] |
X.J. Xie, G. Mistlberger, E. Bakker, J. Am. Chem. Soc. 134 (2012) 16929-16932. DOI:10.1021/ja307037z |
| [4] |
S.L. Wan, Y. Zheng, J. Shen, W.T. Yang, M. Yin, ACS Appl. Mater. Interfaces 6 (2014) 19515-19519. DOI:10.1021/am506641t |
| [5] |
R.X. Zhang, P.F. Li, W.J. Zhang, N. Li, N. Zhao, J. Mater. Chem. C 4 (2016) 10479-10485. |
| [6] |
K. Umezawa, D. Citterio, K. Suzuki, Anal. Sci. 30 (2014) 1348-2246. |
| [7] |
Y. Ma, Y. Zeng, H. Liang, et al., J. Mater. Chem. C 3 (2015) 11850-11856. DOI:10.1039/C5TC03327F |
| [8] |
T. Kowada, H. Maeda, K. Kikuchi, Chem. Soc. Rev. 44 (2015) 4953-4972. DOI:10.1039/C5CS00030K |
| [9] |
H. Lu, Y. Zheng, X. Zhao, et al., Angew. Chem. Int. Ed. 55 (2016) 155-159. DOI:10.1002/anie.201507031 |
| [10] |
S. Leng, Q.L. Qiao, Y. Gao, et al., Chin. Chem. Lett. 28 (2017) 1911-1915. DOI:10.1016/j.cclet.2017.03.034 |
| [11] |
Z.Q. Xu, J.H. Chen, L.L. Hu, et al., Chin. Chem. Lett. 28 (2017) 1935-1942. DOI:10.1016/j.cclet.2017.07.018 |
| [12] |
Y.H. Chen, T.W. Wei, Z.J. Zhang, et al., Chin. Chem. Lett. 28 (2017) 1957-1960. DOI:10.1016/j.cclet.2017.05.010 |
| [13] |
H.B. Sun, S.J. Liu, W.P. Lin, et al., Nat. Commun. 5 (2014) 3601-3609. DOI:10.1038/ncomms4601 |
| [14] |
J.C.G. Bünzli, S.V. Eliseeva, Chem. Sci. 4 (2013) 1939-1949. DOI:10.1039/c3sc22126a |
| [15] |
Y. Sagara, T. Mutai, I. Yoshikawa, K. Araki, J. Am. Chem. Soc. 129 (2007) 1520-1521. DOI:10.1021/ja0677362 |
| [16] |
G.Q. Zhang, J.W. Lu, M. Sabat, C.L. Fraser, J. Am. Chem. Soc. 132 (2010) 2160-2162. DOI:10.1021/ja9097719 |
| [17] |
Y. Gong, G. Chen, Q. Peng, et al., Adv. Mater. 27 (2015) 6195-6201. DOI:10.1002/adma.201502442 |
| [18] |
M. Jin, T.S. Chung, T. Seki, H. Ito, M.A. Garcia-Garibay, J. Am. Chem. Soc. 139 (2017) 18115-18121. DOI:10.1021/jacs.7b11316 |
| [19] |
A.N. Woodward, J.M. Kolesar, S.R. Hall, et al., J. Am. Chem. Soc. 139 (2017) 8467-8473. DOI:10.1021/jacs.7b01005 |
| [20] |
S.Y. Park, Y. Kubota, K. Funabiki, M. Shiro, M. Matsui, Tetrahedron Lett. 50 (2009) 1131-1135. DOI:10.1016/j.tetlet.2008.12.081 |
| [21] |
G. Qian, Z.Y. Wang, Chem.-Asian J. 5 (2010) 1006-1029. DOI:10.1002/asia.200900596 |
| [22] |
L. Cheng, C. Wang, L.Z. Feng, K. Yang, Z. Liu, Chem. Rev. 114 (2014) 10869-10939. DOI:10.1021/cr400532z |
| [23] |
P. Zhang, Z.Q. Guo, C.X. Yan, W.H. Zhu, Chin. Chem. Lett. 28 (2017) 1952-1956. DOI:10.1016/j.cclet.2017.08.038 |
| [24] |
D. Wu, Y.Z. Shen, J.H. Chen, et al., Chin. Chem. Lett. 28 (2017) 1979-1982. DOI:10.1016/j.cclet.2017.07.004 |
| [25] |
F.L. Song, R. Liang, J. Deng, Z.W. Liu, X.J. Peng, Chin. Chem. Lett. 28 (2017) 1997-2000. DOI:10.1016/j.cclet.2017.08.023 |
| [26] |
J.D. Luo, Z.L. Xie, J.W.Y. Lam, et al., Chem. Commun. (2001) 1740-1741. |
| [27] |
B.C. Lin, C.P. Cheng, Z.Q. You, C.P. Hsu, Phys. Chem. Chem. Phys. 13 (2011) 20704-20713. DOI:10.1039/c1cp22535a |
| [28] |
F. Ciardelli, G. Ruggeri, A. Pucci, Chem. Soc. Rev. 42 (2013) 857-870. DOI:10.1039/C2CS35414D |
| [29] |
Z. Mao, Z. Yang, Y. Mu, et al., Angew. Chem. Int. Ed. 54 (2015) 6270-6273. DOI:10.1002/anie.201500426 |
| [30] |
Q. Xiao, J. Zheng, M. Li, et al., Inorg. Chem. 53 (2014) 11604-11615. DOI:10.1021/ic5016687 |
| [31] |
T. Seki, N. Tokodai, S. Omagari, et al., J. Am. Chem. Soc. 139 (2017) 6514-6517. DOI:10.1021/jacs.7b00587 |
| [32] |
J.H. Chen, D.Y. Li, W.J. Chi, et al., Chem.-Eur. J. 24 (2018) 3671-3676. DOI:10.1002/chem.201705780 |
| [33] |
K.P. Guo, F. Zhang, S. Guo, et al., Chem. Commun. 53 (2017) 1309-1312. DOI:10.1039/C6CC09186E |
| [34] |
W. Yang, C.L. Liu, S. Lu, et al., ChemistrySelect 2 (2017) 9215-9221. DOI:10.1002/slct.201701997 |
| [35] |
X. Cheng, D. Li, Z.Y. Zhang, H.Y. Zhang, Y. Wang, Org. Lett. 16 (2014) 880-883. DOI:10.1021/ol403639n |
| [36] |
M. Tanioka, S. Kamino, A. Muranaka, et al., J. Am. Chem. Soc. 137 (2015) 6436-6439. DOI:10.1021/jacs.5b00877 |
| [37] |
H.W. Wu, P. Zhao, X. Li, et al., ACS Appl. Mater. Interfaces 9 (2017) 3865-3872. DOI:10.1021/acsami.6b15939 |
| [38] |
Z.W. Liu, K. Zhang, Q. Sun, et al., J. Mater. Chem. C 6 (2018) 1377-1383. DOI:10.1039/C7TC04698G |
| [39] |
F.S. Zhang, L. Zhai, S.W. Feng, et al., Dyes Pigm. 156 (2018) 140-148. DOI:10.1016/j.dyepig.2018.03.069 |
| [40] |
S.Z. Mo, Q.T. Meng, S.L. Wan, et al., Adv. Funct. Mater. 27 (2017) 1701210. DOI:10.1002/adfm.v27.28 |
| [41] |
H.C. Zhu, J.Y. Huang, L. Kong, Y.P. Tian, J.X. Yang, Dyes Pigm. 151 (2018) 140-148. DOI:10.1016/j.dyepig.2017.12.053 |
| [42] |
K. Jeong, S. Park, Y.D. Lee, et al., Adv. Mater. 25 (2013) 5574-5580. DOI:10.1002/adma.201301901 |
| [43] |
W. Li, X.P. Gan, D. Liu, et al., RSC Adv. 6 (2016) 44599-44605. DOI:10.1039/C6RA05699G |
| [44] |
F. Hu, M.J. Cao, X. Ma, S.H. Liu, J. Yin, J. Org. Chem. 80 (2015) 7830-7835. DOI:10.1021/acs.joc.5b01466 |
| [45] |
H.Y. Zhuo, H.F. Su, Z.Z. Cao, et al., Chem.-Eur. J. 22 (2016) 17619-17626. DOI:10.1002/chem.v22.49 |
| [46] |
M.J. Lin, B. Fimmel, K. Radacki, F. Würthner, Angew. Chem. Int. Ed. 50 (2011) 10847-10850. DOI:10.1002/anie.v50.46 |
| [47] |
G. Qian, J. Qi, J.A. Davey, J.S. Wright, Z.Y. Wang, Chem. Mater. 24 (2012) 2364-2372. DOI:10.1021/cm300938s |
| [48] |
K. Cai, Q.F. Yan, D.H. Zhao, Chem. Sci. 3 (2012) 3175-3182. DOI:10.1039/c2sc21142d |
| [49] |
X. Zhang, B. Li, Z.H. Chen, Z.N. Chen, J. Mater. Chem. 22 (2012) 11427-11441. DOI:10.1039/c2jm30394a |
| [50] |
S.F. Xue, Y.J. Wu, Y.S. Lu, et al., J. Mater. Chem. C 5 (2017) 11700-11707. DOI:10.1039/C7TC04171C |