 2018, Vol. 29
2018, Vol. 29 



b University of Chinese Academy of Sciences, Beijing 100049, China
Enantioseparation is important for racemic compounds, especially for many pharmaceuticals, mainly due to their different pharmacological activities and/or toxicities for the two enantiomers of the same compound in biological environment [1]. Various kinds of chromatographic techniques, such as high performance liquid chromatography, gas chromatography, thin layer chromatography, supercritical fluid chromatography, and capillary electrophoresis can be used to detect enantiomers. Among them, HPLC is the most versatile technique for enantiomeric separation. Although numerous chiral selectors have been reported in HPLC, those based on cyclodextrins (CDs) are still attracting research interests. The truncated cone structures of native CDs enable them to form inclusion complexes with a variety of molecules and the natural chirality of CDs is beneficial for chiral separation. Additionally, derivations of hydroxyl groups at C-6 position, C-2 and C-3 positions can remarkably improve enantioselectivity. CDs and their derivatives can be used as chiral additives [2-5] as well as chiral stationary phases (CSPs) [6-8].
Carboxymethyl-β-cyclodextrin (CM-β-CD) as one of the common β-CD derivatives used for chiral mobile phase additive (CMPA) has reached many successful resolutions for chiral drugs [9-13]. However, addition of such chiral selector into mobile phase may suffer from some drawbacks such as the limiting application in normal phase HPLC or reversed phase HPLC with low aqueous composition because of its poor solubility in these mobile phases. In addition, a relatively low chiral recognition is generally found in the CMPA technique unless a large amount of this additive is applied. In order to overcome these problems, CM-β-CD can be bonded onto silica support as a CSP.
Up to now, there are few reports on the application of CM-β-CD CSP. Park and coworkers reported preparation and use of CM-β-CD-coated zirconia as a CSP, where CM-β-CD is just physically coated on zirconia not chemically bonded [14]. Leonelli et al. reported to use Orpak CDBS-453 to resolve Wieland-Miescher ketone and derivatives [15], where Orpak CDBS-453 was noted to have silica (5 μm) bonded with CM-β-CD as the packing material. Actually, this column is produced by Shodex, a Japanese chromatographic column manufacturer and the Shodex website (http://shodexhplc.com/product-category/l45/) clearly shows that Orpak CDBS-453 is a R, S-hydroxypropyl ether β-cyclodextrin column not a CM-β-CD column. In addition, there is not any report on chemically bonded CM-β-CD CSP.
In the present work, a chiral stationary phase is synthesized by chemically bonding CM-β-CD onto the surface of aminized silica gel through amidation reaction and confirmed by Fourier transform infrared spectroscopy (FT-IR), element analysis (EA) and thermal gravimetry analysis (TGA). Chiral recognition abilities are evaluated with some racemic compounds under reversedphase conditions.
FT-IR spectra were performed on a Thermo Fisher Nicolet 6700 FT-IR Spectrometer (Thermo Fisher, U.S.A.). EA was obtained with a vario MICRO cube instrument (Elementar, Germany). High resolution mass spectrometry (HRMS) was carried out on a microTOF-Q Ⅱ comprising a high-performance hybrid quadrupole time-of-flight mass spectrometer (Bruker, Germany). TGA was recorded on a TG/DSC 2 thermal analyzer (Mettler Toledo, Switzerland). Evaluation of the new column was made on a Shimadzu (Kyoto, Japan) LC-Solution equipped with a LC-20AT binary LC pump, a Shimadzu SPD-20A UV–vis detector and a 7725i manual injector (Rheodyne Inc., U.S.A.) with a 20 μL sample loop. All solvents and reagents used in synthesis and mobile phases were commercially available and directly used without further purification.
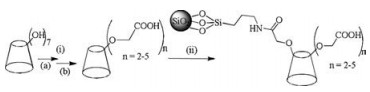
The synthetic route is shown in Scheme 1.

|
Download:
|
| Scheme 1. Synthesis of CM-β-CD CSP. Reagents and conditions: (i) (a) chloroacetic acid/sodium hydroxide/H2O/60 ℃, (b) hydrochloric acid; (ii) EDCI/NHS/3-aminopropyl silica gel/H2O. | |
Chloroacetic acid aqueous solution (17%, 9 mL) was added dropwise to a solution of β-CD (5.00 g) in aqueous 20% sodium hydroxide solution (15 mL) on a water bath (60 ℃) over 10 min. The resulting mixture was stirred for 4 h. Then hydrochloric acid was added to adjust the pH to 6.0-7.0 while the reaction solution was cooled to room temperature. The obtained product was precipitated by addition of excess MeOH. After filtration, the well white precipitate was washed with 3 × 30 mL aqueous 50% MeOH solution and dried in vacuo at 60 ℃ to afford 3.32 g CM-β-CD. FT-IR (curve B, Fig. S1 in Supporting information) (KBr, cm-1): 3388.9, 2928.6, 1602.4, 1416.8, 1329.9, 1079.9, 1030.5, 579.5; HRMS (Fig. S2 in Supporting information) (m/z, negative): 1249.3291, 1271.2849, 1307.3363, 1329.3159, 1365.3399, 1387.3190, 1409.2992 and 1489.2831. The FT-IR spectrum indicates that CM-β-CD exists in the form of sodium CM-β-CD. Two strong peaks at 1602.4 cm-1 and 1416.8 cm-1 are assigned to the asymmetrical stretching vibration and symmetrical stretching vibration of carboxylic salts, respectively, which are not observed in native β-CD. The HRMS results further corroborate the success of the synthesis procedure. Two peaks at 1249.3291 and 1271.2849 mean that two hydroxyl groups on β-CD reacted with chloroacetic acid; Peaks at 1307.3363, 1329.3159 indicate that three hydroxyl groups on β-CD participated in reaction; Peaks at 1365.3399, 1387.3190, 1409.2992 suggest tetrasubstituted β-CD, and 1489.2831 indicates five. The average substitution degree of CM-β-CD is calculated to be 3 according to the HRMS results.
This new CSP was obtained by chemically bonding CM-β-CD onto 3-aminopropyl silica gel. The CM-β-CD (3.32 g), 1-ethyl-3-(3- dimethylaminopropyl)carbodiimide hydrochloride (EDCI) (3.51 g), and N-hydroxysuccinimide (NHS) (2.03 g) were dissolved in distilled water (70 mL), after which Platisil NH2 silica gel (3- aminopropyl silica gel) (1.55 g) was added at 0 ℃ and the reaction was allowed to proceed for 24 h at room temperature, filtrated, washed with water, MeOH, and then dried in vacuo at 60 ℃ for 4 h. CM-β-CD CSP was obtained as white powder (1.94 g, 125%). EA found (%): C 13.02, H 2.68, N 2.93; FT-IR (curve C, Fig. S1 in Supporting information) (KBr, cm-1): 3442.6, 2934.8, 1647.0, 1560.7, 1100.1, 468.5; TGA (curve b, Fig. S3 in Supporting information): Weight loss was 29.9% (w/w). EA shows an increase of carbon content from 7.0% to 13.0%. Surface coverage of CM-β-CD on aminized silica gel was estimated to be about 0.28 μmol/m2 using the following formula [16]:

|
where %C is the increment of carbon content (6.02), nc is the number of carbon atoms in the bonded moiety (48), M is the molecular weight of the bonded moiety (1309) and S is the aminized silica gel special surface (440 m2/g). The FT-IR data shows a new intense absorption peak at 1647.0 cm-1 due to the amide group of the modified silica gel. The loss of weight for CM-β-CD CSP was determined to be 29.9% from 35 ℃ to 800 ℃ under air atmosphere at heating rate of 15 ℃/min, while the loss of weight for aminized silica gel was only 9.6%, which could also confirm the successful bonding of the CM-β-CD CSP.
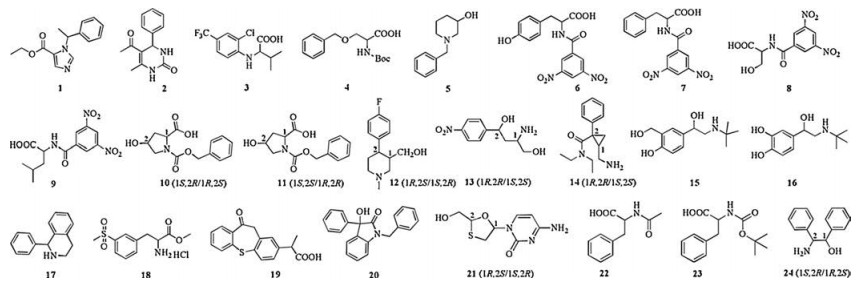
The CSP was slurry packed into stainless steel column (150 mm × 4.6 mm i.d.) using a slurry packing method with an Alltech 1666 HPLC Slurry Packer. Carbon tetrachloride/1, 4-dioxane = 2/1 (v/v) used as the slurry solvent and MeOH as the packing solvent. Chromatographic performances of this CSP were evaluated with 24 racemic compounds listed in Fig. 1 which were either supplied from pharmaceutical companies or prepared in our laboratory (compounds 6–9, Supporting information). All the chromatograms were obtained at 1.00 mL/min and 254 nm unless otherwise specified under reversed-phase mode. Mobile phases were filtered with 0.45 μm membranes and degassed with sonication before use. All the working samples were prepared in MeOH at a concentration of about 1.0–2.0 mg/mL, and the injected volume was 3 μL.

|
Download:
|
| Fig. 1. Structures of chiral analytes used in this study. | |
In order to investigate the effect of mobile phase (salts, pH and organic modifiers) on chiral separation of CM-β-CD CSP, several compounds were chosen for detailed study. Introduction of salt in the mobile phases could greatly improve separation efficiency and peak shape [17]. The influence of ammonium acetate (NH4Ac) concentration on the chiral separation was investigated using a neutral (compound 2), an acidic (compound 7) and a basic (compound 17) compound as model analytes (Table 1). As shown in Table 1, in the absence of NH4Ac in mobile phase, the neutral and acidic compounds could be eluted while the basic compound could not be eluted within 30 min. This is because there is an ion-pairing formation between the protonated basic analyte and the free carboxyl on the CM-β-CD CSP. However, this interaction could not be aroused for the neutral and acidic compounds. It can be noted that the retention factors (k) for all the three analytes are reduced with the amount of NH4Ac in the mobile phase increasing from 0.50% (w/v, 0.0649 mol/L) to 2.00% (w/v, 0.260 mol/L), especially for compound 17. Meanwhile, the selectivity factors (α) for all of them are kept almost unchanged but the resolutions (Rs) decreases slightly. The suitable amount of NH4Ac is 0.50%.
|
|
Table 1 Effect of salt on resolution of compounds 7, 17 and 2 on CM-β-CD CSP.a |

In addition to NH4Ac, ammonium chloride (NH4Cl) and monoammonium phosphate (NH4H2PO4) were tested as other two ammonium salt additives, at identical salt concentration (0.0649 mol/L) and MeOH content varied from 50% to 20% (Tables S1–S3 in Supporting information), the representative chromatograms are depicted in Fig. S4 (Supporting information). The results indicate that anion species also affect the retention and the enantioselectivity. NH4H2PO4 is found to be more effective in reducing retention factors than NH4Cl for the three compounds. However, NH4Ac has different effect in retention for different natural compound. Acidic compound shows the lowest retention in aqueous NH4Ac while basic compound exhibits the highest retention. It's not surprising because different salt solution has different pH which might vary ionization of solute molecule with acidic or basic functional groups. Among these salt additives, NH4Cl produces the highest selectivity for compound 7, NH4Ac for compound 17, and nearly no difference for compound 2. Taking retention and resolution into account, NH4Ac was selected for the following work.
The effects of salt can be explained by Machida's theory which is suitable for most of chiral stationary phases containing a hydrophobic cavity such as crown ether [18], calixarene [19] and vancomycin [20]. Machida proposed that the salt added into the mobile phases affected the enantioselectivity in two aspects. One is that the salt competes against solute for forming inclusion complex which could result in the decrease of retention with salt's amount. The other is ion-pair formation between solute cations and counter anions in mobile phase which might lead to increase in retention with salt's amount. Though competition seems to be dominant for different nature of compounds, the observed results are probably ascribed to the sum of the two mechanisms.
The influences of pH values were summarized in Table S4 (Supporting information) with MeOH-0.50% NH4Ac (30/70, v/v) as the mobile phase. Three acids (acetic acid (HAc), formic acid (FA) and trifluoroacetic acid (TFA)) and three amines (ammonium hydroxide (NH3·H2O), triethylamine (TEA) and diethylamine (DEA)) were used to adjust the buffer pH from 4.0 to 7.5. Results show that the pH value has different effects on retention for different natural analytes. Though a slight difference of k values are observed using different acidic and basic additives for the same analytes, the change tendency is the same. The retention for acidic compound decreases with the increase of pH value, while opposite retention tendency is observed for basic compound. However, the pH has little influence on neutral compound. This may be because pH could alter the ionization state of both acidic and basic compounds which are pH dependent. The acidic and basic compounds show the similar retention behavior to ion-suppression reversed-phase HPLC [21, 22]. In a typical ion-suppression reversed-phase HPLC, the retention for acidic compound shows the reversed S-shaped behavior while the basic compound shows the S-shaped behavior. The enantioselectivity for the acidic and basic compounds are greatly affected by the mobile phase pH. For neutral compound, the enantioselectivity does not vary much with different pH values. As seen in Table S4, a better chiral separation for acidic and basic compound can be obtained in weak acidic and weak basic mobile phase, respectively.
The composition of mobile phase also plays an important role in enantioseparation, not only in retention, but also in selectivity and resolution [23, 24]. The influences of MeOH content in retention can be explained as below. For one aspect, the higher content of MeOH will weaken the hydrophobic interaction between the solutes and CD. For another, increasing MeOH content may attenuate the hydrogen bonding or dipole interactions. Several compounds were selected as the model test analytes, and the data are depicted in Fig. S5 (Supporting information). Each of the analytes can reach the best enantioseparation under a proper composition of mobile phase. For most of them, the retention factor and resolution decrease obviously while the selectivity seldom changes or even does not vary with the increase of MeOH content.
However, not all the compounds follow this general rule and some exceptions exist. Take compound 11 as an example, the retention for the initial enantiomer increases when the MeOH content is increased from 0% to 30%. For the selectivity and resolution, there is a downward trend with this variation. The best resolution reaches up to 9.56 which is suitable for preparative chromatography. The chiral separation can be explicated by Fujimura's competitive inclusion model [25]. According to Fujimura's model for enantiodiscrimination, the aromatic group and the side chain with a chiral carbon compete with each other for CD cavity, and the chiral discrimination is beneficial if the chiral side chain is included into the cavity. Compound 11 contains a phenyl group and a chiral N-heterocyclic side chain, the inclusion formation occurs at the chiral N-heterocyclic ring rather than the aromatic group. The success of enantioseparation is dependent on the interactions of the inclusion formation and the hydrogen bond or the dipole interactions, where the latter two exist on the polar groups of the analyte and the hydroxyl groups on the rim of CD cavity. Another example is for compound 18. The retention, separation factor and resolution for the first diastereoisomer all increase with the MeOH content increases from 60% to 80%, and the highest resolution reaches 6.70. The results imply that increasing the content of MeOH could lead to better enantioseparation and enhanced retention for the first diastereoisomer. However, although the resolution is high, the retention for compound 18 is poor. This can be ascribed to the presence of sulfonyl group which prohibits the phenyl group penetrating into the CD cavity because of its steric hindrance interaction.
To further investigate the effect of organic modifiers, other two protic solvents ethanol (EtOH) and 2-propanol (iPrOH) were used and compounds 2, 7 and 17 were selected as model analytes (Tables S5–S7 in Supporting information). Because of the increasing mobile phase viscosity with the alcohol chain length, the flow was 1.00 mL/min, 0.70mL/min and 0.50 mL/min for MeOH, EtOH and iPrOH, respectively. These results indicate that the elution strength of these organic modifiers decreases in the order of iPrOH > EtOH > MeOH. It is found that only compound 17 gets a slight enantioseparation in the 20% iPrOH-0.50% NH4Ac with the iPrOH varying from 50% to 20%. EtOH is more effective in enantioselectivity than iPrOH but poorer than MeOH. There are no or slight enantioseparations for these compounds with the mobile phase composition in the range of 50/50 (v/v) to 30/70 (v/v) EtOH-0.50% NH4Ac. Compared with 20% MeOH, 20% EtOH seems to obtain a little better enantioselectivity for compounds 2, 7 and a slightly inferior resolution for compound 17.
Acetonitrile (MeCN) as an aprotic organic modifier was also selected for more detailed study in this work. The percentage of MeCN is ranged from 50% to 10% and the amount of NH4Ac in the mobile phase is increasing from 0 to 2.00% (w/v, 0.260 mol/L) (Tables S8–S11 in Supporting information). The results show that 0.50% NH4Ac is also the most suitable amount for the MeCN system. As shown in Table S5, MeOH-0.50% NH4Ac (30/70, v/v) gives a satisfactory enantioseparation for compounds 7, 17 and 2. Nevertheless, the same composition of MeCN-0.50% NH4Ac (30/70, v/v) cannot achieve separation for compounds 7 and 2 but it can lead to partial separation for compound 17. Meanwhile, the retention factors for them are too small. This can be attributed to the lower polarity of MeCN that could be easier to compete with the analytes in occupying the CD hydrophobic cavity leading to stronger elution. So MeCN could not rival MeOH in resolution.
Table 2 shows that CM-β-CD CSP exhibits excellent enantioselectivity for analytes with MeOH-0.50% NH4Ac in appropriate proportions, and most of them have achieved the baseline resolution (Rs > 1.5). Amongst the 24 compounds, there are 9 compounds with chiral center on ring structures and 15 compounds with chiral center on a side chain. For the first 9 compounds, six compounds have reached the baseline resolution, two achieve partial separation and one could not be separated. For the latter 15 compounds, the number of full separation, partial separation and no separation are 6, 4 and 5, respectively. The results demonstrate that a better resolution can be obtained if the chiral center is part of a ring structure rather than on an aliphatic side chain in general. Armstrong and co-workers reported the same conclusion [26]. It was also reported that more rigid analyte led to a bigger energy difference in the formation of inclusion complexes [27]. Consequently, better enantioseparation could be obtained for more rigid analytes. The chiral center on a ring structure is relatively more rigid than on an aliphatic side chain, so a greater energy difference results in the diastereomeric inclusion complexes of the same chiral compound leading to a better resolution.
|
|
Table 2 Enantioseparation of chiral compounds on CM-β-CD CSP and native β-CD bonded CSP-CYCLOBOND I 2000 in the reversed-phase mode. |

{kind=link}
{kind=link}
However, an effective enantioseparation can also be obtained if compounds contain a chiral center on the aliphatic pendant chain with proper substituted groups. Compounds 6 (Rs = 2.20) and 7 (Rs = 1.69) have gained good enantioseparations in 30/70 (v/v) of MeOH-0.50% NH4Ac mobile phase, however, no enantioseparations are observed for compounds 8, 9, 22 and 23 under the same conditions. The data for 23 is not shown because it cannot be eluted within 40 min. Comparing the retention of compound 7 with those of compounds 22 and 23, it could deduce that the retention for these compounds mainly depends on the amino protecting groups. Comparing the resolutions for compounds 6, 7, 8 and 9, it is reasonable that phenyl ring is essential for chiral resolution. Compounds 3 (Rs = 0.95) and 4 (Rs = 0.66) contain both a chiral aliphatic side chain and a phenyl ring but they do not lead to satisfactory resolutions. For compound 4, the phenyl ring is far away from the chiral center making the CSP hard to provide sufficient chiral discrimination. For compound 3, the substituted groups of the phenyl ring are too large to wholly penetrate into the CD cavity. The results indicate that good enantioseparation could be obtained if a compound contains both a chiral side chain and an aromatic group which is close to the chiral center and suitable for CD cavity.
The structural difference between drug compounds 15 and 16 lies in one of the substituent groups on benzene ring, which is hydroxymethyl and hydroxyl, respectively, leading to different chromatograph performances. Compound 16 has a stronger retention and better resolution than compound 15 which could not even be resolved. It is probably due to a weak ionization for phenol hydroxyl that could form greater hydrogen bond and dipole interactions. Compounds 11 and 10 are a pair of cis-trans isomerisms, the surprising separation results should be attributable to the bulky steric hindrance of N-heterocyclic ring for compound 10 hampering the chiral moiety interact with CD cavity.
Table 2 also shows that CM-β-CD CSP exhibits better enenatioseparation than CYCLOBOND I 2000 (produced by Sigma-Aldrich, 150 mm × 4.6 mm i.d.). Most of the analytes can be effectively separated on CM-β-CD CSP and partial of them achieve the baseline separation. However, only three analytes 5, 11 and 18 can be enantioseparated on CYCLOBOND I 2000. The effect of MeOH content for the commercially available column is like the CM-β-CD CSP, the decrease of MeOH content is also helpful in increasing retention and enantiomeric separation for most compounds. The best resolution values for compounds 5, 11 and 18 are 2.31, 1.82 and 0.77, respectively, while the resolution values are 7.01, 9.56 and 6.70 under the same mobile phase conditions for CM-β-CD CSP. For most analytes, shorter retention values are observed on CYCLOBOND I 2000 than CM-β-CD CSP or even no retention. The results imply that carboxymethyl plays positive role in chiral discrimination.
The enantiomeric separation for underivatized β-CD mainly depends on inclusion and hydrogen bond interaction between the CD and the chiral compound. In the case of CM-β-CD, the carboxymethyl moiety can provide additional sites for hydrogen bond, dipole or electrostatic interaction. The additional interactions may assist in "immobilizing" the compound in the diastereomeric inclusion complex. The importance of the additional interactions is also demonstrated by some other researchers [28-32] chiral enantioselectivity is greatly enhanced for the derivatized CD. Thus, the excellent chiral discrimination ability is driven by the synergetic effects of the native CD and modified moiety.
In this work, a CSP derived from CM-β-CD has been successfully bonded onto aminized silica gel via amidation reaction in water solution, which was characterized by FT-IR, EA and TGA. The enantioseparation abilities of this CSP were studied in the reversephase mode by various analytes, and excellent chiral resolutions are shown on this CSP with proper composition of mobile phase. The type and content of salt, mobile phase pH, organic modifiers and structures of analytes play important roles in enantioseparation. Compared with commercially available column CYCLOBOND I 2000, CM-β-CD CSP shows better enantioselectivities.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2017.10.039.
| [1] |
J. Caldwell, J. Chromatogr. A 719 (1996) 3-13. DOI:10.1016/0021-9673(95)00465-3 |
| [2] |
M. Huang, X.F. Zhang, Y. Wang, et al., Chin. Chem. Lett. 24 (2013) 840-844. DOI:10.1016/j.cclet.2013.05.019 |
| [3] |
J.C. Ye, W.Y. Yu, S. Zeng, et al., Biomed. Chromatogr. 24 (2010) 799-807. DOI:10.1002/bmc.v24:8 |
| [4] |
J. Guan, J. Yang, F.M. Li, et al., J. Sep. Sci. 31 (2008) 288-293. DOI:10.1002/jssc.200700369 |
| [5] |
S.L. Ma, S. Shen, N. Grinberg, et al., J. Chromatogr. A 1216 (2009) 1232-1240. DOI:10.1016/j.chroma.2008.12.016 |
| [6] |
Y.P. Zhang, X.M. Liang, A.W. Lei, et al., J. Chromatogr. A 1191 (2008) 188-192. DOI:10.1016/j.chroma.2007.11.018 |
| [7] |
X.P. Chen, H. Yuan, Z.M. Zhou, et al., Chin. Chem. Lett. 19 (2008) 797-800. DOI:10.1016/j.cclet.2008.04.045 |
| [8] |
Q.J. Peng, J.J. Yang, Chin. Chem. Lett. 25 (2014) 1416-1418. DOI:10.1016/j.cclet.2014.05.052 |
| [9] |
T.G. Lu, M.J. Yang, Chin. J. Chromatogr. 25 (2007) 830-833. |
| [10] |
S.M. Shuang, M.M.F. Choi, J. Chromatogr. A 919 (2001) 321-329. DOI:10.1016/S0021-9673(01)00810-X |
| [11] |
M.H. Zhai, L.Q. Su, Y.J. Sun, Chem. Res. Appl. 24 (2012) 450-453. |
| [12] |
Y.J. Shi, C.H. Huo, B.J. Xu, et al., J. Chromatogr. A 1072 (2005) 279-282. DOI:10.1016/j.chroma.2005.03.027 |
| [13] |
C. Roussel, A. Favrou, J. Chromatogr. A 704 (1995) 67-74. DOI:10.1016/0021-9673(95)00166-K |
| [14] |
S.Y. Park, J.K. Park, J.H. Park, et al., Microchem. J. 70 (2001) 179-185. DOI:10.1016/S0026-265X(01)00129-1 |
| [15] |
F. Leonelli, B. Garofalo, M. Sinibaldi, et al., J. Liq. Chromatogr. Related Technol. 26 (2003) 409-424. DOI:10.1081/JLC-120017179 |
| [16] |
A. Berthod, C.D. Chang, D.W. Amstrong, Talanta 40 (1993) 1367-1373. DOI:10.1016/0039-9140(93)80212-A |
| [17] |
L. Chen, L.F. Zhang, S.C. Ng, et al., J. Chromatogr. A 950 (2002) 65-74. DOI:10.1016/S0021-9673(02)00043-2 |
| [18] |
Y. Machida, H. Nishi, K. Nakamura, J. Chromatogr. A 830 (1999) 311-320. DOI:10.1016/S0021-9673(98)00896-6 |
| [19] |
H. Hashem, T. Jira, J. Chromatogr. A 1133 (2006) 69-75. DOI:10.1016/j.chroma.2006.07.074 |
| [20] |
H. Hashem, C. Trundelberg, T. Jira, et al., J. Chromatogr. A 1218 (2011) 6727-6731. DOI:10.1016/j.chroma.2011.07.090 |
| [21] |
P. Nikitas, A. Pappa-Louisi, J. Chromatogr. A 971 (2002) 47-60. DOI:10.1016/S0021-9673(02)00965-2 |
| [22] |
R. LoBrutto, A. Jones, Y.V. Kazakevich, et al., J. Chromatogr. A 913 (2001) 173-187. DOI:10.1016/S0021-9673(00)01012-8 |
| [23] |
R. Sardella, F. Ianni, B. Natalini, et al., Biomed. Chromatogr. 28 (2014) 159-167. DOI:10.1002/bmc.3015 |
| [24] |
Y. Wang, T.T. Ong, S.C. Ng, et al., J. Chromatogr. A 1216 (2009) 2388-2393. DOI:10.1016/j.chroma.2009.01.039 |
| [25] |
K. Fujimura, S. Suzuki, K. Hayashi, et al., Anal. Chem. 62 (1990) 2198-2205. DOI:10.1021/ac00219a009 |
| [26] |
A. Berthod, J.D. Duncan, D.W. Armstrong, et al., J. Pharm. Biomed. Anal. 8 (1990) 123-130. DOI:10.1016/0731-7085(90)80018-K |
| [27] |
M. Rekharsky, Y. Inoue, J. Am. Chem. Soc. 122 (2000) 4418-4435. DOI:10.1021/ja9921118 |
| [28] |
Q.F. Huang, Y. Inoue, C. Yang, et al., J. Org. Chem. 81 (2016) 3430-3434. DOI:10.1021/acs.joc.6b00130 |
| [29] |
Z.Q. Yan, Q.F. Huang, C. Yang, et al., Org. Lett. 19 (2017) 898-901. DOI:10.1021/acs.orglett.7b00057 |
| [30] |
D. Zhao, Y. Chen, Y. Liu, Chin. Chem. Lett. 26 (2015) 829-833. DOI:10.1016/j.cclet.2014.11.028 |
| [31] |
J.C. Gui, Z.Q. Yan, C. Yang, et al., Chin. Chem. Lett. 27 (2016) 1017-1021. DOI:10.1016/j.cclet.2016.04.021 |
| [32] |
A.M. Stalcup, S.C. Chang, D.W. Armstrong, J. Chromatogr. 513 (1990) 181-194. DOI:10.1016/S0021-9673(01)89435-8 |